NOTE: The protocol can be paused anywhere.
1. Basic Setup
- Obtain a BED, Peaks format, or BigWig7 file containing the data to be input into Genome. The file should have extension name "bed", "broadpeaks""narrowpeaks", or "bigWig" respectively.
NOTE: Zipped versions of these type of files will also work. - Use an internet browser to go to genemo.org. Any operating system capable of running most common internet browsers should be able to use GeNemo.
- Choose which species to search against using the dropdown menu. Currently available species include human and mouse.
- Upload user file using a url or a direct upload. BigWig files only work with the url upload method. BED and Peaks format files work with both methods (wiggle files cannot be uploaded as the main data as of now).
2. Optional Setup
- Provide an email address in the corresponding box in order to receive the search results by email when the search is done.
NOTE: When searching a large part of the genome and/or against a large number of tracks (see below), it is recommended that the user provides his/her email, since the search may take a long time. For example, a 100 megabase search takes around 15 s. A link to the search results will be sent to the email address provided when the search is completed. The link will expire in 7 days after the completion of a search. - Provide a bigwig file or the wiggle display file may be from a url. This display file will not affect the results; it will only be shown alongside the results.
- Specify a search range (including the chromosome and base pair positions) in the corresponding box.
- List the chromosome, start base pair, and end base pair.
- Use 'chrN' for the chromosome format, where 'N' is the chromosome number/letter (1, 2, … X, or Y). For the base pairs, just type in the numbers.
- Include spaces between all three entries, or include a colon (:) between chromosome number and the first base pair, and/or a hyphen between the two base pairs. For example: chr1:1000000-2000000, chr1 1000000 2000000, chr1 1000000-2000000, chr1:1000000 2000000.
NOTE: Steps 2.1 – 2.3 are optional.
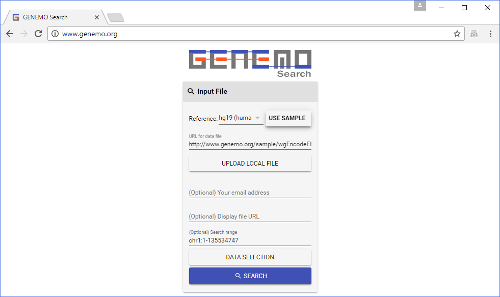
Figure 1: GeNemo's front page with the necessary areas filled out. A user needs to input the species, search file and search range, and select tracks he/she wishes to search against. Email address and display file are optional. Please click here to view a larger version of this figure.
3. Data Selection
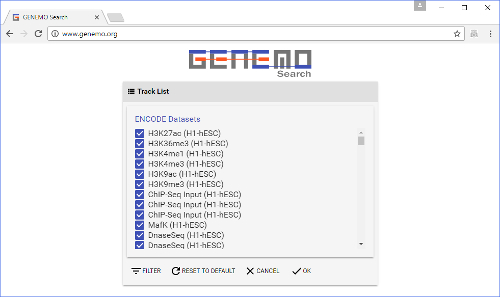
Figure 2: Track selection window. This is brought up by clicking the "DATA SELECTION" button on the front page. Here, users select tracks to search the input file against. Some of the tracks are already selected by default. Please click here to view a larger version of this figure.
- After clicking the data selection button, choose which types of tracks to search against (i.e., to add to the query). The track collection includes many different datasets from labs around the world.
- As the list of tracks is quite long, users may want to use the filter button (on the top) to facilitate track selections. Tracks may be filtered by Experiment, Tissue, Cell Line and/or Lab.
- There are five buttons on the bottom to help execute track selection: Select All, Select None, Add, Filter, Exclude.
- Select All" and "Select None" are self-explanatory.
- The "Add" button adds currently selected tracks to the query. It serves as the logic gate "OR". Note that selecting the filter(s) above (e.g., certain Experiments, Tissues, Cell Lines or Labs) does not automatically add corresponding tracks to the search query. Users must first select tracks (e.g., brain, liver under tissue), and then click the "Add" button to add them to the query. When selecting tracks, note that only the filters specified in the opened tab in the filter window will be applied to the search query. Selections on other tabs will be saved in the filter window, but not applied to the search query.
- The "Filter" button retains only the types of tracks currently selected in the filter window in the query and removes all other types of tracks. It serves as the logic gate "AND". Essentially, "Filter" allows the selection of the interaction between two categories of tracks (e.g., certain tissues with certain Labs). Note that "Filter" does not add the selected types of tracks to the query if they are not already in the query.
- The "Exclude" button removes all types of tracks that are currently selected in the filter window from the query. It serves as the logic gate "NOT", in opposition to the "Filter" function. Again, "Exclude" does not add any tracks currently not selected in the filter window to the query.
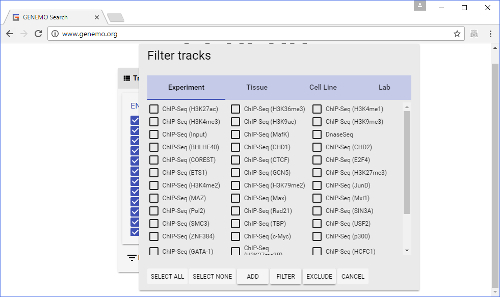
Figure 3: Filter window. This is brought up by clicking the "FILTER" button on the Track selection window. Here, users can select many tracks at the same time, with relative ease. Please click here to view a larger version of this figure.
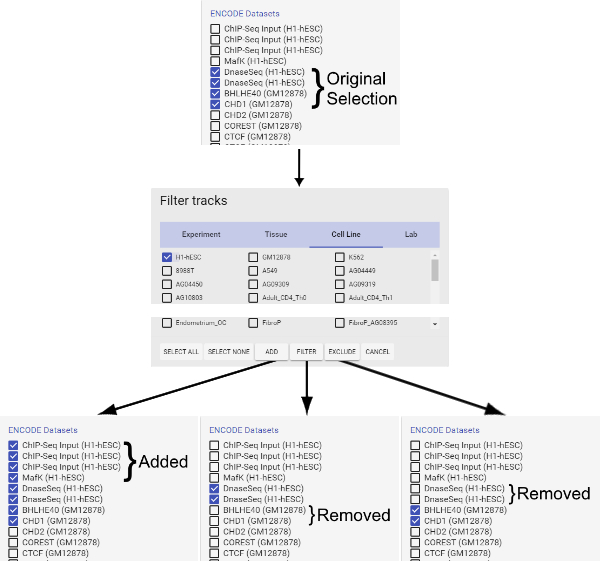
Figure 4: How to use the filter function. Please click here to view a larger version of this figure.
- After adding the desired tracks to the query, click the "Update" button on the bottom right. This is needed in order to accommodate two ways to select data: selecting individual data tracks or filtering/excluding. The "Reset View" button resets the query to the default tracks related to gene expression regulation in human/mouse embryonic stem cells.
NOTE: Selecting tracks to be searched against via "Data selection" is optional but recommended because the default search tracks are most likely not suited to the user's needs.
4. Search and Results
- Click the "Search" button after data selection. The search may take some time.
- Once the search is completed, users will see various boxes in the Results page. Each box represents a section of the genome where a user's data file has a closely matched pattern with one or more of the tracks the user has queried.
- If there are no boxes visible, try searching more types of tracks or making the search range bigger with the same input file. An easy way to do this without redoing everything is clicking the "☰" button next to the logo. This will open up a sidebar that allows the user to modify the search.
- The results may be exported as a BED file by clicking on the "DOWNLOAD BED FILE" button on the bottom of the Results page.
- Click the Visualize button on the top right of each box to visualize the results.
- In the Visualization panel on the right, multiple things are displayed including the data, which incorporates the user input file, the display file if one was inputted, matching tracks, and some default tracks. From the results, the user may compare known ENCODE datasets against the provided dataset for further investigation. The user may also refer to UCSC genes to see the context of the query results. If tracks from multiple cell lines/tissues are selected, the user may use such results to gain insights about the tissue specificity of the similarities between the given dataset and ENCODE datasets.
- On the Results page, user may drag on any tracks to move upstream or downstream of the genome; when the mouse cursor is on the coordinates, the user may use the mouse wheel and/or zoom in and out.
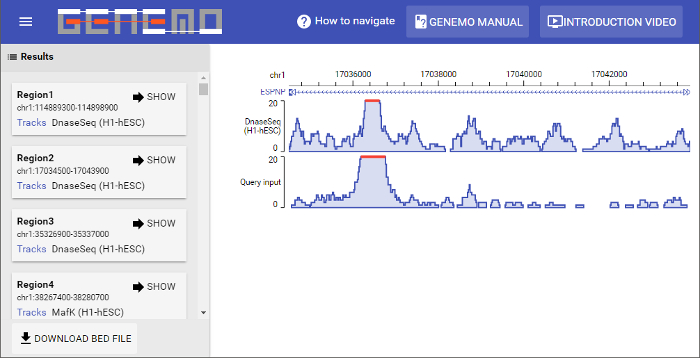
Figure 5: Results page. This particular search returned 363 matching regions. Displaying the first matching region can be done by clicking the "SHOW" button on the bottom left of each resulting region box. On the left part of the display window it can be seen that the two data files (input and selected track) are similar in signal strength pattern. Please click here to view a larger version of this figure.
