Fonte: Jonathan F. Blaize1, Elizabeth Suter1, e Christopher P. Corbo1
1 Departamento de Ciências Biológicas, Wagner College, 1 Campus Road, Staten Island NY, 10301
A avaliação quantitativa dos procariotes pode ser onerosa dada a sua abundância, propensão à proliferação exponencial, diversidade de espécies dentro de uma população e necessidades fisiológicas específicas. Compondo esse desafio, é a natureza de quatro fases em que as bactérias se replicam (lag, log, estacionário e morte). A capacidade de estimar com precisão a concentração de microrganismos é necessária para uma identificação, isolamento, cultivo e caracterização bem-sucedidas (6). Como tal, os microbiologistas têm utilizado diluição serial e várias técnicas de chapeamento por mais de um século para quantificar de forma confiável a carga bacteriana e viral em ambientes clínicos, industriais, farmacêuticos e acadêmicos (2,4,6). Descrições dessa metodologia apareceram pela primeira vez em 1883, quando o cientista e médico alemão Robert Koch publicou seu trabalho sobre agentes causadores de doenças infecciosas (2). Muitas vezes referida como o pai da bacteriologia moderna, as técnicas forenamed de Koch tornaram-se o padrão ouro para a enumeração de microrganismos, culturable ou não, em todo o mundo.
A diluição serial é uma redução sistemática de uma entidade conhecida ou desconhecida (um soluto, organismo, etc.) através da sucessiva re-suspensão de uma solução inicial (solução0) em volumes fixos de um diluente líquido (em branco). Esses espaços em branco geralmente consistem em 0,45% salino, embora a composição possa ser variada (7). Embora um experimentador possa escolher qualquer volume para cada diluído, é na maioria das vezes um múltiplo de 10, facilitando a redução logarítmica da amostra. Por exemplo, a solução0 contém um total de 100 células E. coli suspensas em 10 mL de caldo de nutrientes. Se 1 mL de solução0 for removido e adicionado a 9 mL de soro fisiológico (diluente1),a nova solução (solução1) conteria 1/10 da concentração inicial de E. coli. Neste exemplo, a nova solução (solução1) conteria 10 células E. coli. Repetir este processo removendo 1 mL da solução1 e adicionando-o a outros 9 mL de soro fisiológico (diluente2) renderia a solução 2 , contendoapenasuma única célula E. coli. Uma vez que cada nova solução (9 mL de diluente + 1 mL de solução) contém um total de 10mL, podemos concluir que o fator de diluição para essa redução é de 10 ou que esta foi uma diluição serial de 10 vezes(Figura 1). Como só começamos com 100 células neste exemplo e estamos diluindo por um fator de 10, apenas dois passos são necessários para alcançar a concentração mínima absoluta de 1 célula.
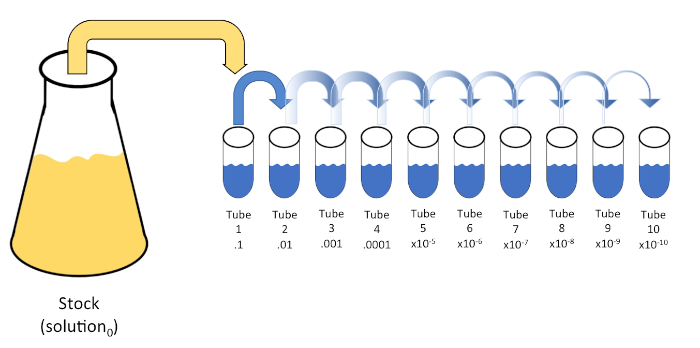
Figura 1: Diluição em série de uma solução de estoque. Uma alíquota de 1 mL da solução de estoque (solução0) é adicionada ao tubo 1 que contém 9 mL de 0,45% de soro fisiológico (dilent1); o produto desta mistura é a solução1. Repita aliquoting 1 mL da solução recém-criada1 e adicioná-lo ao tubo 2. A aliquotação e a resuspensão continuam desta forma até que o tubo final seja atingido, diluindo a concentração de estoque por um fator de 10 cada um a cada etapa. Clique aqui para ver uma versão maior desta figura.
A diluição serial é a técnica mais simples para obter concentrações gerenciáveis de um organismo desejado e é complementada por listras e espalhamento de placas de petri, apenas duas das muitas técnicas de chapeamento usadas por microbiologistas. Este benefício desta abordagem é que o experimentador pode colher cepas puras de uma única espécie ou separar cepas de uma população mista (7). O streaking é realizado introduzindo um organismo a um meio sólido (geralmente consistindo de agarose) que ele crescerá sobre se os nutrientes apropriados estiverem disponíveis. Varrer suavemente um laço inoculante estéril através do meio (de modo que uma raia sutil permanece) em um rígido padrão sinusoidal irá distribuir o organismo proporcionalmente à frequência da forma de onda do experimentador. Dividir a placa de Petri em terços (raia do quadrante) e diminuir a frequência de cada raia à medida que uma nova região da placa é inserida reduzirá gradualmente o número de microrganismos que podem ocupar aquela região, produzindo colônias únicas em vez de um gramado bacteriano inquantificável. O revestimento disseminador não dilui adicionalmente as amostras; um espalhador de vidro estéril é usado para distribuir uma alíquota de mídia de suspensão através de uma placa de petri inteira(Figura 2). As colônias que crescem na placa de propagação surgem de uma única célula e cada colônia no prato pode ser contada para estimar o número de unidades formadoras de colônias por mililitro (CFU) em uma determinada suspensão, representadas como CFU/mL (6)(Figura 3) Ágar macio e trinagem são variações das técnicas acima mencionadas e permitem o isolamento de bacteriófago e triagem mutante, respectivamente (1,7).
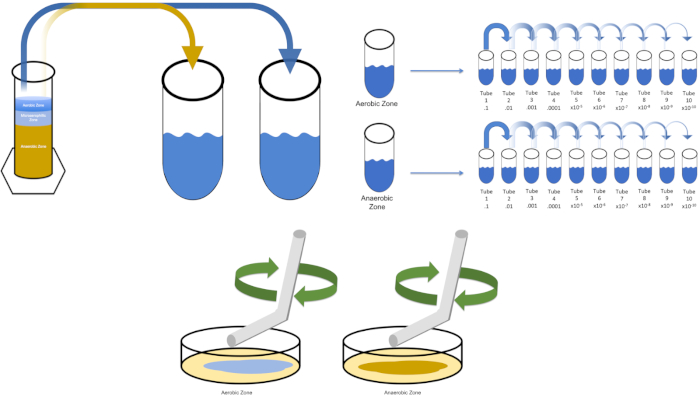
Figura 2: Listramento de placa para enumeração bacteriana e isolamento da tensão. Rotule a parte inferior de uma placa de petri com informações de identificação (nome da bactéria, data, mídia) e divida em terços. Depois de selecionar uma diluição apropriada da amostra de estoque, pegue um laço inoculante estéril (descartável ou inflamado) e submerse-o no tubo de ensaio (aqui, T3). Levante ligeiramente a tampa da placa de petri de um lado para que apenas o laço inoculante possa acessar o ágar. Deslize o laço inoculante através do topo da mídia de uma forma em zig-zag tomando cuidado para não comprometer o ágar. Gire a placa por aproximadamente 1/3rd (~118°) e reduza a frequência do movimento zig-zag. Gire um tempo final e reduza a frequência em zig-zag mais uma vez. Clique aqui para ver uma versão maior desta figura.
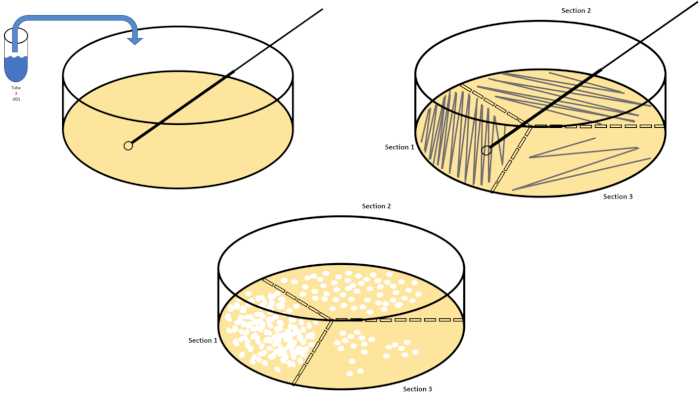
Figura 3: Espalhe o revestimento. 1 g da zona aeróbica foi resuspended em T1 e, em seguida, diluído em série. Um vidro estéril ou vara descartável de plástico é usado para distribuir inóculo em cada prato. Isso se repetiu com 1 g da zona anaeróbica. Clique aqui para ver uma versão maior desta figura.
Como acontece com as diluições seriais, uma escala logarítmica é empregada para expressar a concentração do organismo. O número de colônias cultivadas em placas de petri padrão medindo 100mm x15mm pode ser enumerado manualmente (ou automatizado com o auxílio do processamento computacional) identificando clusters isolados de crescimento. Contagem que totalizam menos de 30 ou mais de 300 devem ser definidas como muito poucos para contar (TFTC) ou muito numerosos para contar (TNTC), respectivamente. No caso deste último, uma diluição seriada deve ser realizada para reduzir a concentração antes de reestilar uma nova placa de petri. A média do número de colônias independentes identificadas a partir de três placas de petri separadas e multiplicando a média pelo fator de diluição produzirá UFC/mL; plotar o registro10 de UFC/mL contra o tempo revelará o tempo médio de geração do organismo (7).