Source: Jonathan F. Blaize1, Elizabeth Suter1, and Christopher P. Corbo1
1 Department of Biological Sciences, Wagner College, 1 Campus Road, Staten Island NY, 10301
Quantitative assessment of prokaryotes can be onerous given their abundance, propensity for exponential proliferation, species diversity within a population, and specific physiological needs. Compounding this challenge, is the four-phase nature in which bacteria replicate (lag, log, stationary and death). The ability to accurately estimate microorganism concentration is necessary for successful identification, isolation, cultivation, and characterization (6). As such, microbiologists have employed serial dilution and various plating techniques for over a century to reliably quantify bacterial and viral load in clinical, industrial, pharmaceutical, and academic laboratory environments (2,4,6). Descriptions of this methodology first appeared in 1883 when the German scientist and physician Robert Koch published his work on infectious disease-causing agents (2). Often referred to as the father of modern bacteriology, Koch's forenamed techniques have become the gold standard for enumeration of microorganisms, culturable or otherwise, throughout the world.
Serial dilution is a systematic reduction of a known or unknown entity (a solute, organism, etc.) through successive re-suspension of an initial solution (solution0) into fixed volumes of a liquid diluent (blanks). These blanks usually consist of 0.45% saline, though the composition can be varied (7). While an experimenter can choose any volume for each diluent, it is most often a multiple of 10, facilitating logarithmic reduction of the sample. For example, solution0 contains a total of 100 E. coli cells suspended in 10 mL of nutrient broth. If 1 mL of solution0 is removed and added to 9 mL of saline (diluent1), the new solution (solution1) would contain 1/10th of the initial concentration of E. coli. In this example, the new solution (solution1) would contain 10 E. coli cells. Repeating this process by removing 1 mL of solution1 and adding it to another 9 mL of saline (diluent2) would yield solution2, containing only a single E. coli cell. Since each new solution (9 mL of diluent + 1 mL of solution) contains a total of 10mL, we can conclude that the dilution factor for this reduction is 10 or that this was a 10-fold serial dilution (Figure 1). Since we only began with 100 cells in this example and we are diluting by a factor of 10, only two steps are required to reach the absolute minimum concentration of 1 cell.
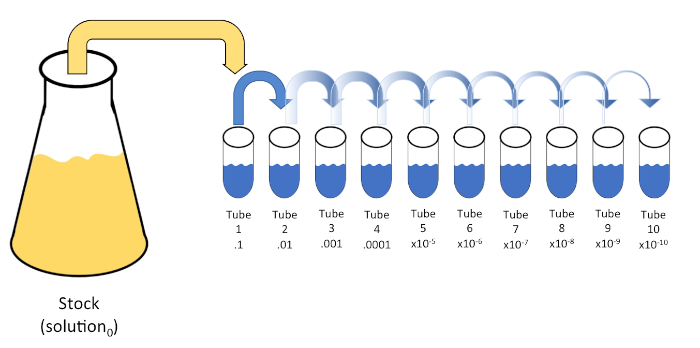
Figure 1: Serial dilution of a stock solution. A 1 mL aliquot of the stock solution (solution0) is added to tube 1 which contains 9 mL of 0.45% saline (dilent1); the product of this mixture is solution1. Repeat by aliquoting 1 mL of the newly created solution1 and adding it to tube 2. Aliquoting and resuspension continues in this fashion until the final tube is reached, diluting the stock concentration by a factor of 10 each with each step. Please click here to view a larger version of this figure.
Serial dilution is the simplest technique for obtaining manageable concentrations of a desired organism and it is complemented by petri dish streaking and spreading, just two of many plating techniques used by microbiologists. This benefit of this approach is that the experimenter can harvest pure strains of a single species or separate strains from a mixed population (7). Streaking is accomplished by introducing an organism to a solid medium (generally consisting of agarose) that it will grow upon if the appropriate nutrients are available. Gently sweeping a sterile inoculating loop across the medium (so that a subtle streak remains) in a rigid sinusoidal pattern will distribute the organism proportionally to the frequency of the experimenter's waveform. Dividing the Petri dish into thirds or fourths (quadrant streak) and decreasing the frequency of each streak as a new region of the dish is entered will gradually reduce the number of microorganisms that can occupy that region, producing single colonies instead of an unquantifiable bacterial lawn. Spread plating does not additionally dilute samples; a sterile glass spreader is used to distribute an aliquot of suspension media across an entire petri dish (Figure 2). The colonies that grow on the spread plate arise from a single cell and each colony on the dish can be counted to estimate the number of colony forming units per milliliter (CFU) in a given suspension, represented as CFU/mL (6) (Figure 3) Soft agar and replica plating are variations of the aforementioned techniques and allow for isolation of bacteriophage and mutant screening, respectively (1,7).
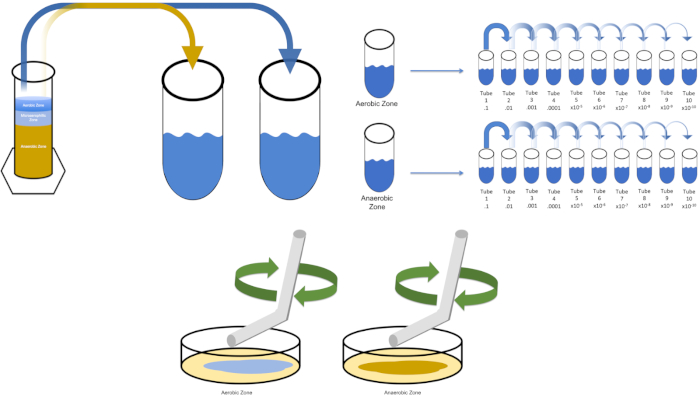
Figure 2: Plate streaking for bacterial enumeration and strain isolation. Label the bottom of a petri dish with identification information (bacteria name, date, media) and divide into thirds. After selecting an appropriate dilution of the stock sample, take a sterile (disposable or flamed) inoculating loop and submerge it the test tube (here, T3). Slightly raise the petri dish cover on one side so that only the inoculating loop can access the agar. Glide the inoculating loop across the top of the media in a zig-zag fashion being careful not to compromise the agar. Rotate the plate by roughly 1/3rd (~118°) and reduce the frequency of zig-zag motion. Rotate a final time and reduce zig-zag frequency once more. Please click here to view a larger version of this figure.
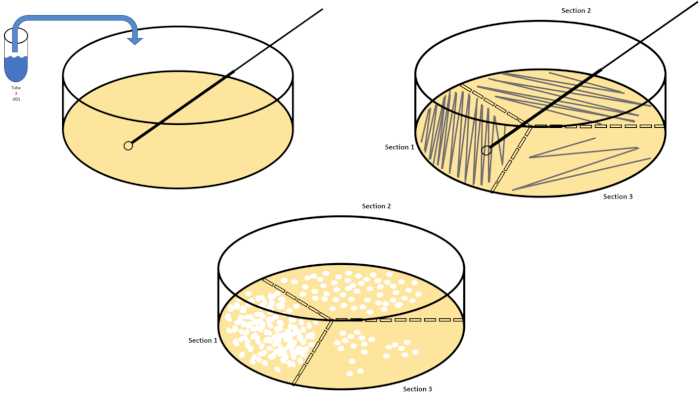
Figure 3: Spread plating. 1 g of the aerobic zone was resuspended in T1 and then serially diluted. A sterile glass or plastic disposable spreading rod is used to distribute inoculum throughout each dish. This was repeated with 1 g of the anaerobic zone. Please click here to view a larger version of this figure.
As with serial dilutions, a logarithmic scale is employed to express organismal concentration. The number of colonies grown in standard petri dishes measuring 100mm x15mm can be enumerated manually (or automated with the aid of computational processing) by identifying isolated clusters of growth. Counts that total fewer than 30 or greater than 300 should be defined as too few to count (TFTC) or too numerous to count (TNTC), respectively. In the case of the latter, a serial dilution should be performed to reduce concentration before restreaking a new petri dish. Averaging the number of self-contained colonies identified from three separate petri dishes and multiplying the mean by the dilution factor will yield CFU/mL; plotting the log10 of CFU/mL against time will reveal the mean generation time of the organism (7).