MultiBac System-Based Purification and Biophysical Characterization of Human Myosin-7a
Summary
This protocol details the procedures for recombinantly producing the human myosin-7a holoenzyme using the MultiBac Baculovirus system and for studying its motility using a tailored in vitro filament gliding assay.
Abstract
Myosin-7a is an actin-based motor protein vital for auditory and visual processes. Mutations in myosin-7a lead to Usher syndrome type 1, the most common and severe form of deaf-blindness in humans. It is hypothesized that myosin-7a forms a transmembrane adhesion complex with other Usher proteins, essential for the structural-functional integrity of photoreceptor and cochlear hair cells. However, due to the challenges in obtaining pure, intact protein, the exact functional mechanisms of human myosin-7a remain elusive, with limited structural and biomechanical studies available. Recent studies have shown that mammalian myosin-7a is a multimeric motor complex consisting of a heavy chain and three types of light chains: regulatory light chain (RLC), calmodulin, and calmodulin-like protein 4 (CALML4). Unlike calmodulin, CALML4 does not bind to calcium ions. Both the calcium-sensitive, and insensitive calmodulins are critical for mammalian myosin-7a for proper fine-tuning of its mechanical properties. Here, we describe a detailed method to produce recombinant human myosin-7a holoenzyme using the MultiBac Baculovirus protein expression system. This yields milligram quantities of high-purity full-length protein, allowing for its biochemical and biophysical characterization. We further present a protocol for assessing its mechanical and motile properties using tailored in vitro motility assays and fluorescence microscopy. The availability of the intact human myosin-7a protein, along with the detailed functional characterization protocol described here, paves the way for further investigations into the molecular aspects of myosin-7a in vision and hearing.
Introduction
Myosins are molecular motor proteins that interact with actin to drive numerous cellular processes1,2,3,4. Humans possess 12 classes and 39 myosin genes5, which are involved in a wide range of physiological functions, such as muscle contraction6 and sensory processes7. Each myosin molecule is a multimeric complex composed of a heavy chain and light chains. The heavy chain is divided into head, neck, and tail regions. The head contains actin- and nucleotide-binding sites that are responsible for ATP hydrolysis and generating force on actin filaments2. The neck is formed by several α-helical IQ motifs where a specific set of light chains are bound. They together function as a lever arm to amplify the motor's conformational changes into large movements8,9,10. The tail contains class-specific subdomains and plays a regulatory role in tuning myosin's motor activity and mediating interactions with cellular binding partners2,11.
Human myosin-7a, a member of class-7 myosins, is essential for auditory and visual processes12,13. The IQ motifs of human myosin-7a are associated with a unique combination of light chains, including the regulatory light chain (RLC), calmodulin, and calmodulin-like protein 4 (CALML4)14,15,16. Besides stabilizing the lever arm, these light chains regulate the mechanical properties of myosin-7a in response to calcium signaling, a feature that appears to be unique to the mammalian isoform14.
Defects in the gene encoding the myosin-7a heavy chain (MYO7A/USH1B) are responsible for Usher syndrome type 1, the most severe form of combined vision and hearing loss in humans17. Additionally, the light chain gene CALML4, is among the candidate genes mapped to contain the causative allele for USH1H, another variant of type 1 Usher syndrome15,18. In the retina, myosin-7a is expressed in the retinal pigment epithelium and photoreceptor cells13. It has been implicated in the localization of melanosomes in the retinal pigment epithelium (RPE)19 and phagocytosis of photoreceptor outer segment disks by the RPE cells20. In the inner ear, myosin-7a is primarily found in the stereocilia, where it plays a critical role in establishing hair bundles and in gating the mechano-electrical transduction process12,21,22.
While the importance of myosin-7a in sensory cells is well established, its functional mechanisms at the molecular level remain poorly understood. This gap in knowledge is partly due to the challenges in purifying the intact protein, especially the mammalian isoform. Recently, significant progress has been made using the MultiBac system to recombinantly express the complete human myosin-7a holoenzyme14. This advancement has enabled structural and biophysical characterizations of this motor protein, leading to the discovery of several unique properties of human myosin-7a that are specifically adapted for mammalian auditory functions14,23.
The MultiBac system is an advanced baculovirus/insect cell platform specifically designed for the expression of eukaryotic multimeric complexes24,25. A key feature of this system is its ability to host multiple gene expression cassettes, each encoding a subunit of the complex, within a single MultiBac baculovirus. The assembly of the multigene expression cassettes is facilitated through a so-called multiplication module: a homing endonuclease (HE) site and a matching designed BstXI site flanking the multiple cloning sites (MCS). This module enables the iterative assembly of a single expression cassette by restriction/ligation, leveraging the fact that the HE and BstXI restriction sites are eliminated upon their ligation. In this paper, human myosin-7a heavy chain, RLC, calmodulin, and CALML4 are each cloned into the multiplication module within the pACEBac1 vector (Figure 1A), which are then assembled into a multigene expression cassette through the iterative process (Figure 1B). The myosin-7a multigene cassette is integrated into the baculoviral genome (bacmid) through the transposition of the mini-Tn7 element from the pACEBac1 vector to the mini-attTn7 target site in the genome (Figure 1C). Following procedures for bacmid purification, baculovirus production, and amplification (Figure 1D,E), the recombinant myosin-7a MultiBac baculovirus is prepared and can be used for large-scale protein production (Figure 1F). Additionally, the myosin-7a light chains can be produced separately in E. coli and purified using a cleavable His6-SUMO tag26,27,28. The purified light chains are useful for studying their binding dynamics and regulation of myosin-7a.
The purified myosin-7a protein can be subjected to structural, biochemical, and biophysical studies to gain insights into the structural-functional regulation of this motor protein. Additionally, its interactions with the actin network and other binding proteins29 can be examined using a variety of in vitro reconstitution approaches. Findings from these analyses will inform the biophysical properties of this myosin, leading to a mechanistic understanding of how myosin-7a drives the cytoskeletal changes and ultimately shapes the unique morphology and function of sensory cells. In this paper, we detail a workflow for actin filament gliding assay that has been specifically adapted for mammalian myosin-7a. Actin filament gliding assay is a robust in vitro motility assay that quantitatively studies the movement of fluorescent actin filaments propelled by a large number of myosin motors immobilized on a coverslip surface30,31,32. The advantages of this assay include its simplicity of setup, minimal equipment requirements (a wide-field fluorescence microscope equipped with a digital camera), and high reproducibility. Additionally, because the motion of actin filaments is driven by a cluster of immobilized myosin motors, this assay is particularly useful for studying the motility of monomeric myosins such as myosin-7a14,33. The protocols include several modifications, from experimental procedures to imaging analysis, specifically tailored to the unique motile properties of mammalian myosin-7a. With the availability of intact myosin-7a protein and the functional characterization protocol outlined here, this paper lays the groundwork for further investigation into the molecular roles of myosin-7a in both physiological and pathological processes.
Protocol
NOTE: Here we describe a protocol for synthesizing the intact human myosin-7a holoenzyme and characterizing its motility in vitro. This protocol is divided into three sections: first, expressing the human myosin-7a using the MultiBac baculovirus protein expression system; Second, purifying myosin-7a light chains separately using the E.coli His6-SUMO system; and lastly, studying the motility of human myosin-7a using the actin-filament gliding assay.
1. MultiBac system-based myosin-7a complex production
- Creating baculovirus multigene cassette for expressing human myosin-7a complex (16 days)
NOTE: It requires one cloning cycle (4 days) to insert the cDNAs of myosin-7a subunits into the pACEBac1 vectors and three cloning cycles to complete the step-wise ligation of all multiplication modules required for expressing the myosin-7a complex, resulting in a total of 16 days.- Clone the cDNAs encoding the myosin-7a heavy chain (HC), calmodulin, CALML4, and RLC into the pACEBac1 vectors using a commercial kit following the manufacturer's protocols (see Table of Materials; Figure 1A). A FLAG tag is inserted at the C-terminus of the myosin-7a HC to aid in purification.
NOTE: Mammalian myosin-7a is produced as two splicing isoforms, differing only by a segment of 11 amino acids at the N-terminus23. This short N-terminal extension plays a critical role in regulating the ATPase activity of myosin-7a14. It has been shown that adding an N-terminal tag can disrupt the normal regulation by this N-terminal extension and significantly reduce the motor's enzymatic activity and motility14. Therefore, a C-terminal FLAG tag is employed to avoid disrupting the protein's natural regulation. - Construct the multigene expression cassette for the myosin-7a holoenzyme using homing endonuclease/BstXI multiplication as described below (Figure 1B).
NOTE: The homing endonuclease I-CeuI produces cohesive ends that are compatible with the ends generated by the BstXI digest. Upon ligation, a homing endonuclease/BstXI hybrid restriction site is created that cannot be cut by either enzyme. The same procedure can be repeated to integrate more multiplication modules. If multiple restriction sites for I-CeuI and BstXI are present on the insert or vector constructs, these additional sites must be eliminated by site-directed mutagenesis to ensure that the I-CeuI and BstXI cutting sites are unique. - Insert preparation: Digest the insert construct (e.g., pACEBac-M7a HC) with the homing endonuclease I-CeuI (see Table of Materials), and then purify the linearized plasmid using a PCR Purification Kit (see Table of Materials). Further, digest the linearized plasmid with BstXI (see Table of Materials), separate the fragment containing the gene expression cassette using gel electrophoresis, and purify using a Gel Extraction Kit (see Table of Materials).
NOTE: I-CeuI and BstXI require different buffer conditions, according to manufacturer's protocols, to achieve 100% activity. In addition, a complete cut with I-CeuI requires a minimum of 3 h, while BstXI requires only 15 min of incubation time. The digestions thus need to be performed sequentially. - Vector preparation: Linearize the vector construct (e.g., pACEBac-CaM) through BstXI digestion. Alkaline Phosphatase is included in the vector preparation to prevent the vector plasmid from ligating to itself (see Table of Materials) . Obtain the digestion product by electrophoresis and purify it with a Gel Extraction Kit.
- Ligation: Ligate the I-CeuI/BstXI-treated insert and the BstXI-treated vector with T4 DNA Ligase (see Table of Materials). Perform ligation reactions at room temperature (20 °C -25 °C) for 10 min with a 1:1 ratio by mass of the insert DNA to vector DNA, keeping the total mass close to 100 ng in each ligation reaction.
- Transformation and plasmid analysis: Transform the ligation mixtures into DH5α cells (see Table of Materials) following the manufacturer's recommendations and screen the positive colonies for gentamicin resistance. Purify the plasmids using a commercial kit (see Table of Materials), and verify the correct clones (e.g., plasmids containing both M7a HC and CaM) based on specific restriction digestion patterns and DNA sequencing of the inserts.
- Integration of multiple gene expression cassettes: Repeat steps 1.1.2 through 1.1.6 to integrate additional gene cassettes expressing other myosin-7a subunits.
- Clone the cDNAs encoding the myosin-7a heavy chain (HC), calmodulin, CALML4, and RLC into the pACEBac1 vectors using a commercial kit following the manufacturer's protocols (see Table of Materials; Figure 1A). A FLAG tag is inserted at the C-terminus of the myosin-7a HC to aid in purification.
- Recombinant bacmid DNA production and purification (4 days; Figure 1C).
NOTE: Day 1 – Bacterial transformation and growing of transformed colonies. Days 2-3 – Growing colonies and screening for successful transformation. Day 3 – Selecting and streaking positive colonies, starting overnight inoculation. Day 4 – Purification of bacmid DNA.- Transform DH10Bac competent cells (see Table of Materials) with 1 ng of the sequenced pACE-M7aHC-RLC-CALML4-CaM plasmid following the manufacturer's recommendations.
- Plate 20-200 µL of the cells on an agar plate containing 50 µg/mL kanamycin, 7 µg/mL gentamicin, 10 µg/mL tetracycline, 100 µg/mL halogenated indolyl-β-galactoside, and 40 µg/mL IPTG (see Table of Materials) and incubate the plate at 37˚C for 48 h to allow for the development of deep blue color in negative clones.
NOTE: The halogenated indolyl-β-galactoside stains colonies that continue to express LacZ (indicating the transposition was unsuccessful), enabling the selection of white colonies where transposition was successful. - Carefully select an isolated white colony and grow cells overnight in 5 mL of LB medium containing 50 µg/mL kanamycin, 7 µg/mL gentamicin, and 10 µg/mL tetracycline at 37 ˚C in an orbital shaker at 225 rpm.
- Centrifuge the culture at 2,465 x g for 10 min to pellet the E coli. Discard the supernatant and resuspend the pellet in 300 µL of Buffer P1 from the Kit.
NOTE: While buffers from the Miniprep kit are used here, the protocol for purifying the bacmid product varies from the protocol provided with the kit. - Add 300 µL of the Buffer P2 from the Kit. Mix by inverting the tubes a few times, and then incubate at room temperature for 3 min.
- Add 300 µL of the N3 buffer from the Kit and mix by inversion to stop the lysis reaction. Centrifuge at 17,885 x g for 10 min at 4 °C.
- Transfer the supernatant to a clean sterile tube and repeat the centrifugation for another 10 min.
- Transfer 800 µL of the supernatant to another sterile 1.7 mL microcentrifuge tube (see Table of Materials) and add 600 µL of cold isopropanol (see Table of Materials), mixing by rigorous inversion. Then centrifuge at 17,885 x g for 20 min at 4 °C.
- Discard the supernatant and locate the small, white pellet. Wash the pellet with 800 µL of cold 70% ethanol (see Table of Materials) and centrifuge at 17,885 x g for 5 min at 4 °C. Carefully add ethanol along the tube wall to avoid disturbing the pellet.
- Discard the supernatant and repeat the ethanol wash once. Air dry the pellet at room temperature for 5 min. Carefully aspirate any excess liquid, if necessary.
- Dissolve the pellet with 20 µL of EB buffer from the Kit. Ensure the pellet is fully dissolved by flicking the tube or gently mixing by pipetting. Determine concentration using a spectrophotometer (see Table of Materials).
- Adjust the buffer volume as needed to make the bacmid concentration at approximately 1 µg/µL. Store bacmid DNA at 4 °C until ready for P0 virus production.
NOTE: The purified bacmid can be kept at 4 ˚C and remains effective for several months. We have seen successful transfections with bacmid products up to 1 year old. Alternatively, the bacmid can be aliquoted and stored at -20 ˚C. Multiple freeze/thaw cycles should be avoided as they decrease the transfection efficiency.
- Transfect Sf9 insect cells with bacmid to produce recombinant baculovirus (6 days; Figure 1D)
NOTE: Day 1 – Transfection of cells with bacmid DNA. Days 2-6 – Observe growth and expression of cells. Day 6 – Collection of P0 virus.- Add Sf9 cells in culture media (see Table of Materials) to a 6-well plate at a concentration of 0.33 x 106 cells/mL with 3 mL per well. Let the plate sit at room temperature for 30 min to allow the cells to attach to the bottom of the plate.
- For each well, prepare a transfection mixture by combining 10 µL of cationic lipid transfection reagent (see Table of Materials) with 250 µL of improved Minimal Essential Medium (MEM) media (see Table of Materials) in a sterile microcentrifuge tube. Mix by inversion and incubate at room temperature for 5 min.
- Add 1 µg (based on concentration determined in step 1.2.12) of undiluted myosin-7a bacmid to the transfection mixture. Mix by inversion and incubate at room temperature for 5 min.
- Equally distribute the entire DNA-lipid mixture dropwise into the wells. Leave one well for non-transfected cells as a negative control.
- Seal the 6-well plate with film (see Table of Materials) and incubate at 27 °C for 6 days. Observe growth and detachment throughout this time. If there is a fluorescent tag present on the construct, check for fluorescence development (Figure 2).
- When the transfected cells show signs of late-stage infection, transfer the medium containing the virus from infected wells to a 15 mL conical tube and centrifuge at 805 x g for 10 min.
- Transfer the supernatant to a clean 15 mL conical tube, wrap it in aluminum foil, and store it at 4 °C. This product is now the P0 virus and can be stored and used effectively for up to four months from our experience.
- Amplify baculovirus stock (3-5 days ; Figure 1E)
- Prepare 100 mL (or desired volume) of Sf9 cells at a concentration of 2 x 106 cells/mL in Sf9 cell culture media in a 250 mL Erlenmeyer flask (see Table of Materials).
- Add 1: 100 ratio (v/v) of P0 virus stock to the Sf9 cell suspension culture and incubate at 27 ˚C in an orbital shaker at 135 rpm.
- Monitor growth every 24 h until cells achieve ~90% cell death. Once at this range, centrifuge the cell suspension at 500 x g for 10 min and collect the supernatant containing the P1 virus.
- Filter the supernatant using 0.22 µm vacuum filtration (see Table of Materials). Keep the filtered product in the dark and store it at 4 °C. This product is now the P1 virus.
NOTE: Virus titer decreases with long-term storage at 4 ˚C. Consider making fresh virus stock if it has been stored for more than 4 months, or titer the virus stock before use in expression34.
- Protein Expression (60 -72 h between beginning the expression and collection of the cell pellet ; Figure 1E)
- Prepare log-phase growing Sf9 cells at a concentration of 2 x 106 cells/mL and add P1 virus at a ratio of 12 mL of virus to 300 mL of cell suspension.
NOTE: The protein yield is approximately 1 mg/L. It is recommended to use a minimum of 1.5 L of cell suspension for this protein expression. - Culture the cells at 27 °C in an orbital shaker at 135 rpm for approximately 60 h.
NOTE: Small samples can be collected at different time points and analyzed using SDS gels to assess protein expression levels. Additionally, if the proteins are fused with fluorescent tags, expression levels can be evaluated using fluorescence microscopy. The cells should be harvested no later than the onset of cell death. - Centrifuge the cell suspension at 3,735 x g for 5 min at 4 °C to pellet the cells. The pellets can be stored at -80 ˚C until further use or preserved for long-term storage. We typically use cell pellets within weeks but have recovered active protein several months after freezing.
- Prepare log-phase growing Sf9 cells at a concentration of 2 x 106 cells/mL and add P1 virus at a ratio of 12 mL of virus to 300 mL of cell suspension.
- Protein purification (2 days ; Figure 1F)
NOTE: Day 1 – Protein extraction and purification using FLAG-affinity chromatography. Day 2 – Aliquot and freeze samples after overnight dialysis. The protocol described below is for purifying myosin-7a from 1.5 L cell pellets.- Prepare Master Buffer containing 10 mM MOPS (pH 7.2), 500 mM NaCl, 10 mM MgCl2, 1 mM EGTA, 1 µg/mL leupeptin, and 100 µg/mL phenylmethylsulfonyl fluoride (PMSF). Filter it with a 0.22 µm pore size filter and store the buffer at 4 ˚C (see Table of Materials).
NOTE: PMSF is unstable in aqueous solutions. Prepare the PMSF stock solution as 1 M in 100% ethanol and store it at -20 ˚C for up to 6 months. - Prepare 75 mL of Extraction Buffer by supplementing the Master Buffer with 3 EDTA-free protease inhibitor tablets, 2 mM ATP, and 0.1 mM dithiothreitol (DTT; see Table of Materials).
- Add 75 mL of Extraction Buffer to the pellet while it is still frozen. Leave the pellet in the Extraction Buffer on ice until it thaws. Use a serological pipette to break up the pellet to accelerate the thawing process.
- Using a ½-inch (13 mm) tip, sonicate the resuspension on ice for 10 min at 50% amplitude, 5 s on, 5 s off.
- Centrifuge the total lysate at 33,746 x g for 30 min at 4 °C. During the centrifugation, prepare 1.5 mL of FLAG resin by washing 3 mL of the 50% slurry of Anti-FLAG affinity resin (see Table of Materials) with 40 mL of PBS. Pellet the resin by centrifuging at 800 x g for 2 min at 4 °C. Carefully remove the PBS without disturbing the resin.
- Add the supernatant from the lysate centrifugation to the washed resin and incubate with continuous rotation at 4 °C for 1 h.
- Pellet the resin by centrifuging at 800 x g for 2 min at 4°C. Without disturbing the resin, remove the supernatant.
- Resuspend the resin in 40 mL of Master Buffer and centrifuge at 800 x g for 2 min at 4 °C. Carefully remove the supernatant without disturbing the resin. Repeat the wash 1x.
- Transfer the washed resin to two polypropylene spin columns (see Table of Materials). Wash each column 1x with 2 mL of Master Buffer. Remove the Master Buffer by centrifuging the column at 1,400 x g for 2 min at 4 °C. The resin will be packed on the bottom.
- Prepare an Elution Buffer by adding 100 µg/mL of FLAG-peptide (see Table of Materials) to the Master Buffer.
- Add 300 µL of Elution Buffer to the packed resin in each column and gently mix by pipetting to ensure the resin is completely hydrated by the Elution Buffer. Let the resin incubate with the Elution Buffer on ice for 10 min.
- Centrifuge the columns at 1,400 x g for 2 min at 4 °C. Transfer the flowthrough to a clean microcentrifuge tube and keep it on ice. This is the first fraction.
- Repeat steps 1.6.11-1.6.12 such that 4 fractions are collected from each column.
- Perform SDS-PAGE gel electrophoresis35 with the elution fractions to determine their relative concentrations. While the gel runs, prepare a Dialysis Buffer containing 500 mM NaCl, 10 mM MOPS, 2 mM MgCl2, 0.1 mM EGTA, and 1 mM DTT.
- Combine the most concentrated fractions. Transfer the sample to a 10K MWCO dialysis cassette (see Table of Materials) and dialyze overnight at 4 °C. Use 1 L dialysis buffer for approximately 500 µL of protein. Perform at least three changes of the dialysis buffer.
NOTE: The spin column and centrifugation elution method allow proteins to be eluted at high concentrations (0.5-1 mg/mL). Further concentrating steps are not usually required. However, if more concentrated proteins are desired, load samples onto 100,000 MWCO concentrating tubes (see Table of Materials) and centrifuge at 1,400 x g for 5 min at 4 ˚C. Repeat this process until the desired protein solution volume is achieved. - The next day, carefully unload the sample from the dialysis cassette. Aliquot 15 µL per PCR tube and drop the tubes into a container of liquid nitrogen for flash-freezing. The frozen proteins can be stored at -80 ˚C for future use (Figure 3A).
NOTE: The yield of myosin-7a using this method is usually between 0.5 and 1 mg/L.
- Prepare Master Buffer containing 10 mM MOPS (pH 7.2), 500 mM NaCl, 10 mM MgCl2, 1 mM EGTA, 1 µg/mL leupeptin, and 100 µg/mL phenylmethylsulfonyl fluoride (PMSF). Filter it with a 0.22 µm pore size filter and store the buffer at 4 ˚C (see Table of Materials).
2. Purifying the light chains RLC and CALML4 using the E. coli His6-SUMO system (7 days )
NOTE: Days 1-4 – Cloning and purification of the plasmids. Day 5 – Bacterial transformation. Days 6-7 – Purification, aliquoting, and freezing of final proteins.
- Clone the cDNAs encoding RLC and CALML4 into a pET-SUMO vector, which contains a His6 sequence prior to SUMO to aid in purification. When needed, the His6-SUMO domain can be cleaved from the fusion protein using a SENP2 protease26,36.
- For each light chain protein, transform BL21 E. coli competent cells (see Table of Materials) with the plasmid and start a 5 mL overnight liquid culture with the transformed cells.
- The next day, inoculate the 5 mL overnight culture into 1 L of Luria-Bertani (LB) broth containing 50 µg/mL kanamycin. Allow the culture to grow at 37 ˚C with continuous shaking at 270 rpm until the optical density (O.D.) reaches 0.6 – 1.0.
NOTE: The O.D. value doubles approximately every 20 min once it reaches 0.2. Monitor the O.D. frequently. - Initiate protein expression by adding 250 µM isopropyl b-D-1-thiogalactopyranoside (IPTG) to the culture. Incubate for 3-5 h at 37 ˚C or 16 ˚C overnight with continuous shaking at 270 rpm.
- Pellet the E. coli by centrifuging the cell suspension at 4,347 x g for 15 min at 4 °C. Discard the supernatant.
- Prepare Resuspension Buffer containing 50 mM Tris-HCl, 1 M NaCl, 5 mM Imidazole, and 10% glycerol, pH 7.4 (see Table of Materials). Keep on ice.
- Resuspend the pellet in 15-25 mL of Resuspension Buffer and transfer the suspension to a clean beaker, adjusting the total volume to 35 mL. Add DNase and RNase to the suspension at a 1:1000 (v/v) ratio for a final concentration of 1 µg/mL and 2 µg/mL, respectively. Add one EDTA-free protease inhibitor tablet, 4 mM 2-mercaptoethanol, and 1% Triton X to the suspension (see Table of Materials). Keep on ice for 20 min.
- Sonicate the pellet suspension in ice water with 1 s on, and 1 s off for a total of 1 min at 100% amplitude. Leave the homogenized lysate at 4 ˚C with continuous rotation for 2 h.
NOTE: The addition of Triton X aids in lysing the cells in preparation for sonication. - Centrifuge the lysates at 26,900 x g for 45 min at 4 ˚C. While this is occurring, begin preparing the cobalt resin (see Table of Materials). Use 500 µL of cobalt resin for 1 L of E. coli culture. Wash the resin 2x with ultrapure water and once with Suspension Buffer by centrifuging at 4,347 x g for 2 min at 4 °C.
- Following lysate centrifugation, combine the supernatant with the washed resin and rock gently at 4 ˚C for 30 min.
- Centrifuge the lysate-resin suspension at 4,347 x g for 2 min at 4 °C. The resin will be packed at the bottom of the tube. Without disturbing the resin, remove the supernatant.
- Wash the resin with Resuspension Buffer by centrifuging at 4,347 x g for 2 min at 4 °C. Remove the supernatant. Repeat washes until the supernatant is colorless.
- Prepare Elution Buffer containing 50 mM Tris HCl, 300 mM NaCl, 5 mM Imidazole, 10% glycerol, 4 mM 2-mercaptoethanol, and 0.25 µM SENP2 protease (see Table of Materials). Keep it on ice.
- Resuspend the resin in 1 mL of Resuspension Buffer and incubate it overnight at 4 °C with continuous rotation.
- The next day, centrifuge at 4,347 x g for 2 min to pellet the resin. Collect the supernatant, which contains the cleaved light chain protein and the protease.
- Add 1 mL of Elution Buffer to the resin. Repeat centrifugation and collect the supernatant.
- Remove the protease by fast protein liquid chromatography (FPLC). Briefly, prepare the size exclusion column (see Table of Materials) with a buffer containing 50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, and 5 mM DTT (pH 7.4). Concentrate the supernatant to 500 µL using a 10,000 MWCO concentrating tube. Load the concentrated sample onto a column connected to an automated chromatography system using a syringe, flow through with a buffer containing 50 mM Tris-HCl, 300 mM NaCl, 10% glycerol, and 5 mM DTT (pH 7.4). Monitor the collected fractions using an ultraviolet spectrophotometry. Collect the protein fractions with a molecular weight corresponding to the light chains.
NOTE: The protease can also be removed by affinity chromatography if a purification tag (e.g., GST tag) is engineered into the protease. - Run an SDS-PAGE gel with the elution fractions to determine the purity and relative concentrations of the light chain proteins in each fraction. Desired fractions are those that contain concentrated light chain proteins without any visible protease bands. Combine the desired protein fractions and concentrate them using a 10,000 MWCO concentrating tube35.
- Flash freeze aliquots of protein in liquid nitrogen and immediately transfer to -80 °C for long-term storage (Figure 3B).
NOTE: The yield of RLC and CALML4 using the E. coli His6-SUMO system is about 1-2 mg/L.
3. Myosin-7a-tailored actin filament gliding assay (3 h)
NOTE: The methods used in this section are similar to those described for other myosins31 with the major modifications being the incubation and application of myosin in high ionic strength buffer and the long interval required to achieve accurate measurement of frame-to-frame displacements.
- Prepare 20 µM filamentous actin (F-actin) by polymerizing globular actin (G-actin) in polymerization buffer (50 mM KCl, 25 mM MOPS, 2 mM MgCl2, 1 mM DTT (pH 7.0)) at 4 °C overnight. Label F-actin by adding 1.2x molar excess rhodamine-phalloidin (see Table of Materials). Cover with aluminum foil and incubate on ice for at least 2 h.
NOTE: G-actin can be purchased from commercial vendors (see Table of Materials) or purified from rabbit muscle acetone powder31,37. Labeled F-actin can be stored at 4 °C for up to 2 months at this concentration. - Prepare a flow chamber as described previously31. Briefly, place two strips of double-sided tape 2-3 mm apart on a cleaned and nitrocellulose-treated microscope slide (see Table of Materials). Put the coverslip nitrocellulose side down onto the strips, creating a flow chamber with an approximate volume of 10 µL (Figure 4).
- Performing actin filament gliding assay
- To the flow chamber, add one chamber volume of 0.2 mg/mL purified human myosin-7a protein in 500 mM NaCl Motility Buffer (500 mM NaCl, 20 mM MOPS, 5 mM MgCl2, 0.1 mM EGTA (pH 7.4)). Let incubate for 5 min at room temperature.
NOTE: It is critical to add myosin-7a in a high salt buffer as this relieves the autoinhibited state and allows myosin-7a to adhere to the surface in the activated conformation. - Wash the flow chamber with three-chamber volumes of 1 mg/mL BSA (see Table of Materials) in 150 mM NaCl Motility Buffer (150 mM NaCl, 20 mM MOPS, 5 mM MgCl2, 0.1 mM EGTA, 1 mM DTT (pH 7.4)) drawing the liquid through using a clean wipe at the outlet to absorb excess liquid. Incubate for 1 min after the 3rd wash.
- Flow one chamber volume of 10 nM rhodamine phalloidin-labeled F-actin in 150 mM NaCl Motility Buffer through the flow chamber. Monitor binding of actin filaments to the surface under a fluorescent microscope with 100x objective. The density of actin filaments should be optimal (Figure 5A), ensuring enough filaments in the field-of-view for tracking yet sparse enough to prevent the overlapping of multiple filaments, which complicates tracking.
- Wash the flow chamber with three-chamber volumes of 150 mM NaCl Motility Buffer to remove unbound actin filaments and excess rhodamine-phalloidin.
- Initiate the motility by adding one chamber volume of the final buffer (200 mM NaCl, 20 mM MOPS, 5 mM MgCl2, 0.1 mM EGTA, 50 mM DTT, 2 mM ATP, 2.5 mg/ml glucose, 100 µg/mL glucose oxidase, 2 µM RLC, 2 µM CaM, 2 µM CALML4).
- Record images on a fluorescence microscope using 561 nm excitation to visualize rhodamine phalloidin-labeled actin. Find an area on the slide with the desired actin density (Figure 5A) and capture images every 30 s for 30 min.
NOTE: The velocity of mammalian myosin-7a is approximately 5 nm/s. When setting the acquisition frame rate, it is important to ensure that the displacements of the filaments between frames are long enough (more than one pixel) to allow for accurate tracking. Subpixel movements between frames can cause an overestimation of the speed. The frame rate of 30 s allows the actin filaments to move about 150 nm between frames, corresponding to more than two pixels on the microscope. However, care should be taken that significant displacement can be observed on the microscope being used. If available, multi-position recording can be used to simultaneously collect several field-of-view for analysis.
- To the flow chamber, add one chamber volume of 0.2 mg/mL purified human myosin-7a protein in 500 mM NaCl Motility Buffer (500 mM NaCl, 20 mM MOPS, 5 mM MgCl2, 0.1 mM EGTA (pH 7.4)). Let incubate for 5 min at room temperature.
- Analyzing the movement of the actin filaments using the FAST program (installed on a Mac)38.
NOTE: The FAST program used here is only one of many options for quantifying filament motion. We chose to use FAST because it is automated, fast, and does not require paid software. Other options include MATLAB-based algorithms such as FIESTA, ImageJ/FIJI-based methods such as MTrack2 and Manual Tracking, and Python-based methods such as Philament39.- Download and install the FAST program (github.com/NeilBillington/FASTrack3 – python3 compatible version with an additional module for. nd2 file conversion. NB: Original python2 version can be found at github.com/turalaksel/FASTrack).
- Run the FAST program as described in38 using a tolerance value of 33 to retain smooth moving filaments only.
NOTE: If images contain drift to mechanical issues with the stage or from collecting several series concurrently, the movies should first be stabilized. For this, we recommend the FIJI plugin Image Stabilizer, available for download at the following address – (https://www.cs.cmu.edu/~kangli/code/Image_Stabilizer.html). - Use the FAST program to analyze the newly created .tif images by first setting the -xmax and -ymax values. These set the parameters for the scatter plot output by the program and represent the longest filament length (in nm) and the maximum filament velocity (in nm/s). We used 20,000 nm for -xmax and 20 nm/s for -ymax in this example to ensure all data is captured (Figure 5D).
- Use -px to set the pixel size of the acquired image, which here will be 65 nm, and -minv to set the minimum velocity (in nm/s) required of a filament for inclusion in the analysis. For the low velocity of myosin-7a in this example, use 0.1 nm/s.
- Use -maxd to set the maximum distance allowed between frames for filaments to be linked. This is set to avoid linking separate filaments between frames and in this example, we use the default 2000 nm.
- Use-pt to set the tolerance for fluctuations in filament velocities and only include filaments with smooth movements. This example uses a tolerance threshold of 33%. This means that filaments with a velocity standard deviation larger than 33% of the velocity mean will be excluded from further analysis.
- Lastly, use -d to indicate the folder containing the .tif stacks for analysis. The entire command string with the given parameters and values will be: fast -xmax 20000 -ymax 20 -px 65 -minv 0.1 -maxd 2000 -pt 33 -d [folder name containing movies].
NOTE: The FAST program will output scatterplots (Figure 5D) with their associated raw data, which can be extracted for analysis and visualization in other programs (Figure 5C). It will also output a plot of the paths of the tracked filaments (Figure 5B), which should be used to verify that the tracking is working as expected. Examples of the plots and other possible data visualizations are included below.
Representative Results
The purified myosin-7a complex and light chain proteins can be evaluated by SDS-PAGE gel electrophoresis, as shown in Figure 3. The band above the 200 kDa marker corresponds to the myosin-7a heavy chain (255 kDa). The three bands migrating between the 22 and 14 kDa markers from top to bottom are RLC (20 kDa), calmodulin, and CALML4, respectively. While calmodulin and CALML4 have a similar molecular weight of approximately 17 kDa, the two proteins can be separated using a 16% Tris-Glycine gel.
Video 1 and Figure 5 demonstrate a characteristic actin filament gliding assay with mammalian myosin-7a. An immediate feature revealed by this assay is the slow motility of myosin-7a. The video is played back at 500 times normal speed to better visualize the movement of actin filaments driven by this myosin. This assay can be modified to further study how external factors such as temperature, ionic strength, and solution viscosity influence the activity of myosin-7a. Additionally, myosin-7a binding proteins can be introduced into the flow cell to investigate their effects on actomyosin interactions and motility.

Figure 1: Workflow for MultiBac-system-based myosin-7a holoenzyme production. (A) Insert the cDNAs encoding the myosin-7a heavy chain, RLC, CALML4, and calmodulin into the multiple cloning sites (MCS) of the pACEBac1 vector. (B) Construct the multigene expression cassette for encoding the myosin-7a complex by iteratively ligating the pACEBac1 vectors that contain the cDNAs of the myosin-7a heavy chain, RLC, CALML4, and calmodulin. (C) Integrate the myosin-7a multigene cassette into the baculovirus genome through the transposition of the mini-Tn7 element from the pACEBac1 vector to the mini-attTn7 target site in the genome. (D) Produce baculoviruses expressing the myosin-7a complex by transfecting Sf9 cells with the recombinant bacmid containing the myosin-7a multigene cassette. (E) The baculovirus can be amplified by inoculating the P0 virus into an Sf9 cell suspension culture and collecting the supernatant. The resulting product, the P1 virus, can be used for large-scale protein expression. (F) Workflow for the FLAG-affinity column-based purification of the myosin-7a complex. Please click here to view a larger version of this figure.

Figure 2: Representative fluorescent images of Sf9 cells transfected with bacmid expressing myosin-7a with a C-terminal GFP tag. The images show the normal progression of baculovirus infection: cells begin to express myosin-7a around day 4 post-transfection, with nearly all cells becoming infected within one week. Please click here to view a larger version of this figure.

Figure 3: SDS-gels of purified myosin-7a complex and light chain proteins. (A) Purified full-length human myosin-7a holoenzyme using the MultiBac system. The heavy chain (HC) and light chains are indicated. (B) Purified RLC and CALML4 using the His6-SUMO system. Calmodulin (CaM) is purchased (see Table of Materials) and purified from the Bovine brain. Please click here to view a larger version of this figure.
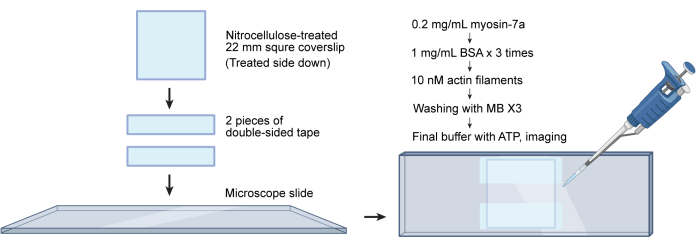
Figure 4: Workflow for flow chamber assembly and actin gliding assays. A nitrocellulose-treated coverslip is attached to a microscope slide using two pieces of double-sided tape. This creates a flow chamber with a volume of approximately 10 µL. Proteins are added sequentially to the chamber, while excess solutions are wicked through the channel using tissue paper. Please click here to view a larger version of this figure.

Figure 5: Representative results of gliding actin filament assays and the tracking analysis. (A) Example frame from a timelapse recording showing actin filament movement driven by surface-decorated myosin-7a motors. (B) Filament tracking image output from the FAST program for the same field-of-view as shown in (A). (C) Representative histogram of the actin gliding velocity generated by mammalian myosin-7a: 4.2 ± 1.4 nm/s (mean ± standard deviation; the number of tracks = 550). (D) Example of autogenerated output from the FAST program showing the calculated filament length and velocities. %STUCK shows the percentage of filaments that are deemed to be stuck based on the user supplied minimum velocity cutoff. MVEL represents the mean velocity of all non-stuck filaments. Please click here to view a larger version of this figure.
Video 1: Actin filament gliding assay performed with myosin-7a. Purified full-length human myosin-7a is attached to the surface. This movie was acquired at 1 frame every 30 s with 200 ms exposure. Please click here to download this Video.
Discussion
Presented here is a detailed protocol for the production of recombinant human myosin-7a protein from insect cells. Although the Sf9/baculovirus system has been used to produce a variety of myosins40,41,42,43, only recently has the mammalian myosin-7a been successfully purified using the MultiBac baculovirus system14. Mammalian myosin-7a is found to associate with three types of light chains, all of which are essential for the protein's structural and functional integrity14. This is in contrast to its invertebrate homolog and most other myosins, which typically bind to only one or two light chain types44,45. This means that to synthesize the human myosin-7a holoenzyme, at least four different genes must be expressed simultaneously in each Sf9 cell. In this case, the MultiBac system offers significant benefits over co-infection with multiple baculoviruses because it ensures reproducible ratios of the myosin-7a complex subunits in each infected cell. In fact, Belyaev et al. have statistically demonstrated this: as the number of virus types increases, the likelihood of cells receiving an equal virus ratio decreases dramatically46. For example, with two virus types, only 12.78% of cells achieve an equal ratio, while this percentage drops to 0.29% when four virus types are used, assuming each virus type is distributed between cells independently. This variability can be problematic when the subunit proteins need to assemble at a consistent ratio to produce the maximum yield of the complex. While this paper focuses on mammalian myosin-7a, it is increasingly evident that the MultiBac system offers a more optimized approach for producing multimeric complexes than the co-infection method, particularly for proteins with more subunits and produced in low yields.
Several factors are critical for achieving high-yield production of myosin-7a protein using this method. First, it is crucial to determine the optimal timing for harvesting Sf9 cells. This allows maximum protein production while minimizing the detrimental effects of cell lysis and death caused by the virus. MultiBac-based protein complex production often exhibits a delayed peak in expression compared to monocistronic baculovirus systems or when expressing smaller proteins. We found that the best time to harvest cells infected with the myosin-7a MultiBac virus is between 60 – 65 h post-infection. Continuous monitoring of protein expression levels throughout the process is strongly recommended. This can be accomplished using fluorescent microscopy if a fluorescent protein tag is fused or through SDS-PAGE analysis otherwise. Additionally, it is important to monitor cell viability concurrently to identify the optimal timing for achieving the highest protein yield with minimal cell death.
Myosin-7a is a large monomeric myosin susceptible to protein degradation14. To prevent degradation during the purification process, it is important to ensure that all buffers are pre-chilled and all procedures are conducted at 4 ˚C. In addition, minimizing the exposure of myosin-7a to proteases is critical. Besides utilizing protease inhibitor cocktails, we recommend keeping the incubation of the cell lysate with FLAG-resin to just 1 h. Extending this duration has not been shown to significantly improve resin binding or increase total protein yield and may pose a higher risk of protein degradation.
A distinctive feature of mammalian myosin-7a, compared to isoforms from lower species44, is its combination of unique light chain components. These light chains play important regulatory roles in the function of myosin-7a. For instance, calmodulin dynamically interacts with the myosin-heavy chain in a Ca2+-dependent manner47, modulating its motility and mechanical output, a mechanism that seems specifically adapted to the mammalian auditory hair cells14. While the exact binding affinities of calmodulin to individual IQ motifs have yet to be determined in the context of the whole molecule, we have observed that some calmodulin gradually dissociates as the excess light chains in the cell lysate are removed during purification. This may alter the mechanical properties of myosin-7a. To mitigate this, we employ spin columns combined with gentle centrifugation for the elution step. This allows proteins to be eluted in a small volume and at a high concentration, which can obviate the need for concentration steps. This practice shortens the total protein purification process and minimizes the risk of light chain dissociation. In actin gliding assays, we include excess light chain proteins in the final buffer to ensure that the native light chains remain bound with the myosin-7a heavy chain.
The requirement for excess light chains represents one of several differences between the actin gliding assay with myosin-7a and that of other well-studied myosins such as myosin-2. Myosin-2 assays are typically performed at low ionic strength (e.g., 50 mM). As ionic strength is raised towards physiological (150 mM), actin filaments tend to detach, and motility slows. In the case of myosin-7a, motility is stalled at low ionic strength, and gliding is only observed at greater than or equal to 150 mM. Due to the autoinhibition of full-length myosin-7a, it is applied to the coverslip in a high ionic strength solution in a manner akin to how myosin filaments are dissolved before application to allow proteins to bind in an orientation and conformation that allows subsequent gliding.
The low velocity observed in actin gliding assays with myosin-7a introduces some challenges in acquiring and analyzing motility data. Indeed, the translocation is several orders of magnitude slower compared to skeletal muscle myosin, which was used when this assay was originally developed30. This low velocity can lead to difficulty in accurate measurement and an overestimation of velocity that scales with frame rate48. The localization precision of any tracking method is finite, and even a static object has an apparent shift in position between successive images. As the sampling rate increases, this leads to an artificially high value for velocity. This effect is true for any type of motility assay, but the relative effect is very small for assays where a large movement of several pixels occurs between frames. By taking data points at very long intervals (every 60 s, 90 s, etc.), it can be verified that the measured velocity is accurate since the value should be unaffected by the sampling rate. Since the recording interval for myosin-7a is so long, the delay can be taken advantage of to record from several positions during the same acquisition. This effectively allows for the recording of several movies at one time. A disadvantage of this method is that mechanical imperfections with the stage will lead to additional drift due to the switching of positions. This can be accounted for using image stabilization as described above.
In the original Python2 version of the FAST software (github.com/turalaksel/FASTrack), each output data point represents a velocity for a filament within an N-frame window (5 frames by default). A modified Python3 version of the software (github.com/NeilBillington/FASTrack3) includes an additional output in which data points are on a filament-by-filament basis. The default plots produced by the software are based on the N window-type dataset. Both output types are equally valid, and although the original output will produce many more data points per movie, we typically use the filament-based data since this is more directly comparable to existing methods for quantifying filament gliding and yields more intuitive information about the numbers of filaments with specific velocities in a particular movie. Note that even in this filament-type analysis, the number of data points will not correspond exactly to the true number of filaments since tracking stops when filaments cross paths, and new tracks are then detected as they emerge after crossing.
Limitations of the methods described in this paper are that mammalian protein is being expressed in insect cells. Although many such proteins, including this one, have been successfully expressed in this way, there are other expression systems that may more closely mimic the native environment of the protein. Mammalian expression systems could introduce important post-translational modifications to the protein which are absent in the insect system. The same is true to an even greater extent for expression in a bacterial system, as used here for producing myosin light chains. Nevertheless, we believe the relative simplicity and high yield in these cases outweigh the potential limitations. The in vitro motility assay that is used to characterize the protein is limited in how much information it can yield about the protein. For example, many aspects of myosin regulation are masked or circumvented by the assay and thus cannot be investigated using this technique. Many other assay types, such as single-molecule motility, optical trapping, and biochemical and biophysical techniques, exist to investigate the properties of myosin in vitro, but the filament gliding assay is chosen here as a method to measure myosin activity because it requires less protein than many biochemical assays, is simple to perform and where successful motility can be demonstrated, tells us that the protein is both biochemically and mechanically active.
The workflow described here represents a series of methods for producing high-quality myosin-7a protein and characterizing its mechanical properties. Although these methods are specifically tailored to this myosin class, the expression and purification procedures are more generally useful as a guideline for how to produce this type of labile protein, consisting of several distinct polypeptides and with a tendency for dissociation and degradation. In addition, the characterization methods are useful for all types of motor proteins, and in particular, the considerations of data acquisition and analysis parameters are intended to be useful for anyone measuring the translocation rate of low-velocity molecular motors.
Divulgaciones
The authors have nothing to disclose.
Acknowledgements
We thank the Microscopy Imaging Facility and Visual Function and Morphology Core at West Virginia University for discussion and help with image analysis. This work is supported by the tenure-track startup funds from West Virginia University School of Medicine to R.L. This work is also supported by National Institute of General Medical Sciences (NIGMS) Visual Sciences Center of Biomedical Research Excellence (Vs-CoBRE) (P20GM144230), and the NIGMS West Virginia Network of Biomedical Research Excellence (WV-INBRE) (P20GM103434).
Materials
| 1.7 mL microcentrifuge tubes | VWR | 87003-294 | |
| 1X FLAG Peptide | GenScript | N/A | Custom peptide synthesis |
| 22x22mm No. 1.5 coverslips | VWR | 48366-227 | |
| 250 mL Conical Centrifuge Tubes | Nunc | 376814 | |
| 250 mL Vented Erlenmyer Shaker Flask | IntelixBio | DBJ-SF250VP | |
| 2-Mercaptoethanol | VWR | M131 | |
| 75x25x1 mm Vistavision microscope slides | VWR | 16004-42 | |
| Actin Protein (>99% Pure) | Cytoskeleton | AKL99 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit | Millipore Sigma | UFC510024 | |
| Amicon Ultra-4 Centrifugal Filter Unit | Millipore Sigma | UFC801024 | |
| ANTI-FLAG M2 Affinity Gel | Millipore Sigma | A2220 | |
| ATP | Millipore Sigma | A7699 | |
| ATP | Millipore Sigma | A7699 | |
| Bio-Spin Disposable Chromatography Column | Bio-Rad | 732-6008 | |
| BL21 Competent E. coli | New England Biolabs | C2530H | |
| Bluo-Gal | Thermo Fisher | 15519028 | |
| Bovine Serum Albumin | Millipore Sigma | 5470 | |
| BstXI Enzyme | New England Biolabs | R0113S | |
| Calmodulin | Millipore Sigma | 208694 | |
| Catalase | Millipore Sigma | C40 | |
| Champion pET-SUMO Expression System | Thermo Fisher | K30001 | |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche Diagnostics | 5056489001 | |
| Cutsmart Buffer | New England Biolabs | B6004S | |
| DL-Dithiothreitol | Millipore Sigma | DO632 | |
| DL-Dithiothreitol | Millipore Sigma | DO632 | |
| DNase I, Spectrum Chemical | Fisher Scientific | 18-610-304 | |
| Double-Sided Tape | Office Depot | 909955 | |
| EGTA, Molecular Biology Grade | Millipore Sigma | 324626-25GM | |
| EGTA, Molecular Biology Grade | Millipore Sigma | 324626-25GM | |
| Ethanol | Thermo Fisher | BP2818 | |
| ExpiFectamine Sf Transfection Reagent | Gibco | A38915 | |
| FAST program | http://spudlab.stanford.edu/fast-for-automatic-motility-measurements; | ||
| Fisherbrand Model 505 Sonic Dismembrator | Fisher Scientific | FB505110 | |
| Gentamicin Reagent Solution | Gibco | 15710-064 | 10 mg/mL in distilled water |
| Glucose | Millipore Sigma | G5767 | |
| Glucose Oxidase | Millipore Sigma | G2133 | |
| Glycerol | Invitrogen | 15514-011 | |
| HisPur Cobalt Resin | Thermo Fisher | 89966 | |
| I-CeuI Enzyme | New England Biolabs | R0699S | |
| Image Stabilizer Plugin | https://www.cs.cmu.edu/~kangli/code/Image_Stabilizer.html | ||
| ImageJ FIJI | https://imagej.net/Fiji/Downloads | ||
| Imidazole | Millipore Sigma | I2399 | |
| In-Fusion Snap Assembly Master Mix | TaKaRa | 638948 | |
| IPTG | Thermo Fisher | 15529019 | |
| Isopropanol | Fisher Scientific | A451SK | |
| Kanamycin | Fisher Scientific | AAJ67354AD | |
| Large Orifice Pipet Tips | Fisher Scientific | 02-707-134 | 1-200uL |
| LB Agar, Ready-Made Powder | Thermo Fisher | J75851-A1 | |
| Leupeptin Protease Inhibitor | Thermo Fisher | 78435 | |
| Magnesium chloride | Thermo Fisher | J61014.=E | 1M |
| Magnesium chloride | Thermo Fisher | J61014.=E | 1M |
| Max Efficiency DH10Bac Competent Cells | Gibco | 10361012 | |
| Microcentrifuge Tubes, 1.7mL | VWR | 87003-294 | |
| Microcentrifuge Tubes, 1.7mL | VWR | 87003-294 | |
| Microcentrifuge Tubes, 1.7mL | VWR | 87003-294 | |
| Microscope | Nikon | Model: Eclipse Ti with H-TIRF system with 100X TIRF objective | |
| Microscope Camera | ORCA-Fusion BT | ||
| Microscope Laser Unit | Andor iXon Ultra | ||
| Miller's LB Broth | Corning | 46-050-CM | |
| MOPS | Millipore Sigma | M3183 | |
| MOPS | Millipore Sigma | M3183 | |
| NanoDrop One/OneC Microvolume UV-Vis Spectrophotometer | Thermo Fisher | ND-ONE-W | |
| NanoDrop One/OneC Microvolume UV-Vis Spectrophotometer | Thermo Fisher | ND-ONE-W | |
| NEB 5-alpha Competent E.coli (High Efficiency) | New England Biolabs | C2987H | |
| NEBuffer r3.1 | New England Biolabs | B6003S | |
| NIS Elements | Nikon | ||
| NIS-Elements | Nikon | ||
| Nitrocellulose | LADD Research Industries | 53152 | |
| Opti-MEM I Reduced Serum Medium | Gibco | 31985070 | |
| pACEBac1 Vector | Geneva Biotech | ||
| Parafilm | Millipore Sigma | P7793 | |
| PMSF | Millipore Sigma | 78830 | |
| PureLink RNase A (20 mg/mL) | Invitrogen | 12091021 | |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 | |
| QIAquick Gel Extraction Kit (50) | QIAGEN | 28704 | |
| QIAquick PCR Purification Kit (50) | QIAGEN | 28104 | |
| Quick CIP | New England Biolabs | M0525S | |
| Rhodamine phalloidin | Invitrogen | R415 | |
| S.O.C. Medium | Invitrogen | 15544034 | |
| SENP2 protease | PMID:17591783 | Purified in the lab | |
| Sf9 cells | Thermo Fisher | 11496015 | |
| Sf-900 III SFM (1X) – Serum Free Media Complete | Gibco | 12658-027 | |
| Slide-A-Lyzer G3 Dialysis Cassettes, 10K MWCO, 3 mL | Thermo Fisher | A52971 | |
| Sodium chloride | Millipore Sigma | S7653 | |
| Sodium chloride | Millipore Sigma | S7653 | |
| Stericup Quick Release Vacuum Driven Disposable Filtration System | Millipore Sigma | S2GPU01RE | |
| Superdex 75 Increase 10/300 GL | Cytiva | 29148721 | |
| T4 DNA Ligase | New England Biolabs | M0202S | |
| T4 DNA Ligase Buffer – 10X with 10mM ATP | New England Biolabs | B0202A | |
| Tetracycline Hydrochloride | Millipore Sigma | T7660-5G | |
| Tris | Millipore Sigma | 10708976001 | |
| Triton X | American Bioanalytical | 9002-93-1 |
Referencias
- Hartman, M. A., Spudich, J. A. The myosin superfamily at a glance. J Cell Sci. 125 (Pt 7), 1627-1632 (2012).
- Sellers, J. R. Myosins: a diverse superfamily. Biochim Biophys Acta. 1496 (1), 3-22 (2000).
- Titus, M. A. Myosin-driven intracellular transport. Cold Spring Harb Perspect Biol. 10 (3), a021972 (2018).
- Quintanilla, M. A., Hammer, J. A., Beach, J. R. Non-muscle myosin 2 at a glance. J Cell Sci. 136 (5), jcs.260890 (2023).
- Peckham, M. Coiled coils and SAH domains in cytoskeletal molecular motors. Biochem Soc Trans. 39 (5), 1142-1148 (2011).
- Batters, C., Veigel, C., Homsher, E., Sellers, J. R. To understand muscle you must take it apart. Front Physiol. 5, 90 (2014).
- Friedman, T. B., Sellers, J. R., Avraham, K. B. Unconventional myosins and the genetics of hearing loss. Am J Med Genet. 89 (3), 147-157 (1999).
- Geeves, M. A., Holmes, K. C. The molecular mechanism of muscle contraction. Adv Protein Chem. 71, 161-193 (2005).
- Batters, C., Veigel, C. Mechanics and activation of unconventional myosins. Traffic. 17 (8), 860-871 (2016).
- Houdusse, A., Sweeney, H. L. How myosin generates force on actin filaments. Trends Biochem Sci. 41 (12), 989-997 (2016).
- Odronitz, F., Kollmar, M. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 8 (9), R196 (2007).
- Moreland, Z. G., Bird, J. E. Myosin motors in sensory hair bundle assembly. Curr Opin Cell Biol. 79, 102132 (2022).
- Williams, D. S., Lopes, V. S. The many different cellular functions of MYO7A in the retina. Biochem Soc Trans. 39 (5), 1207-1210 (2011).
- Hollo, A., et al. Molecular regulatory mechanism of human myosin-7a. J Biol Chem. 299 (10), 105243 (2023).
- Choi, M. S., et al. The small EF-hand protein CALML4 functions as a critical myosin light chain within the intermicrovillar adhesion complex. J Biol Chem. 295 (28), 9281-9296 (2020).
- Ebrahim, S., et al. Stereocilia-staircase spacing is influenced by myosin III motors and their cargos espin-1 and espin-like. Nat Commun. 7, 10833 (2016).
- Whatley, M., et al. Usher syndrome: Genetics and molecular links of hearing loss and directions for therapy. Front Genet. 11, 565216 (2020).
- Ahmed, Z. M., et al. USH1H, a novel locus for type I Usher syndrome, maps to chromosome 15q22-23. Clin Genet. 75 (1), 86-91 (2009).
- Liu, X., Ondek, B., Williams, D. S. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat Genet. 19 (2), 117-118 (1998).
- Gibbs, D., Kitamoto, J., Williams, D. S. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci U S A. 100 (11), 6481-6486 (2003).
- Houdusse, A., Titus, M. A. The many roles of myosins in filopodia, microvilli and stereocilia. Curr Biol. 31 (10), R586-R602 (2021).
- Schwander, M., Kachar, B., Muller, U. Review series: The cell biology of hearing. J Cell Biol. 190 (1), 9-20 (2010).
- Li, S., et al. Myosin-VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nat Commun. 11 (1), 2066 (2020).
- Fitzgerald, D. J., et al. Protein complex expression by using multigene baculoviral vectors. Nat Methods. 3 (12), 1021-1032 (2006).
- Sari, D., et al. The MultiBac baculovirus/insect cell expression vector system for producing complex protein biologics. Adv Exp Med Biol. 896, 199-215 (2016).
- Courtney, K. C., et al. Synaptotagmin-7 outperforms synaptotagmin-1 to promote the formation of large, stable fusion pores via robust membrane penetration. Nat Commun. 14 (1), 7761 (2023).
- Courtney, K. C., et al. The complexin C-terminal amphipathic helix stabilizes the fusion pore open state by sculpting membranes. Nat Struct Mol Biol. 29 (2), 97-107 (2022).
- Courtney, K. C., et al. Synaptotagmin 1 oligomerization via the juxtamembrane linker regulates spontaneous and evoked neurotransmitter release. Proc Natl Acad Sci U S A. 118 (48), e2113859118 (2021).
- Yu, I. M., et al. Myosin 7 and its adaptors link cadherins to actin. Nat Commun. 8, 15864 (2017).
- Kron, S. J., Spudich, J. A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci U S A. 83 (17), 6272-6276 (1986).
- Sellers, J. R. In vitro motility assay to study translocation of actin by myosin. Curr Protoc Cell Biol. Chapter 13, 13.2 (2001).
- Tripathi, A., Bond, C., Sellers, J. R., Billington, N., Takagi, Y. Myosin-specific adaptations of in vitro fluorescence microscopy-based motility assays. J Vis Exp. (168), e62180 (2021).
- Yang, Y., et al. A FERM domain autoregulates Drosophila myosin 7a activity. Proc Natl Acad Sci U S A. 106 (11), 4189-4194 (2009).
- Jorio, H., Tran, R., Kamen, A. Stability of serum-free and purified baculovirus stocks under various storage conditions. Biotechnol Prog. 22 (1), 319-325 (2006).
- JoVE Science Education Database. Separating protein with SDS-PAGE. Basic methods in cellular and molecular biology. , (2023).
- Mikolajczyk, J., et al. Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J Biol Chem. 282 (36), 26217-26224 (2007).
- Pardee, J. D., Spudich, J. A. Purification of muscle actin. Methods Enzymol. 85 Pt B, 164-181 (1982).
- Aksel, T., Choe Yu, E., Sutton, S., Ruppel, K. M., Spudich, J. A. Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep. 11 (6), 910-920 (2015).
- Bowser, R. M., Farman, G. P., Gregorio, C. C. Philament: A filament tracking program to quickly and accurately analyze in vitro motility assays. Biophys Rep. 4 (1), 100147 (2024).
- Bird, J. E., et al. Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking. Proc Natl Acad Sci U S A. 111 (34), 12390-12395 (2014).
- Billington, N., Wang, A., Mao, J., Adelstein, R. S., Sellers, J. R. Characterization of three full-length human nonmuscle myosin II paralogs. J Biol Chem. 288 (46), 33398-33410 (2013).
- Billington, N., et al. Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr Biol. 25 (7), 942-948 (2015).
- Lu, W., et al. Competition between kinesin-1 and myosin-V defines Drosophila posterior determination. Elife. 9, 54216 (2020).
- Liu, R., et al. A binding protein regulates myosin-7a dimerization and actin bundle assembly. Nat Commun. 12 (1), 563 (2021).
- Heissler, S. M., Sellers, J. R. Myosin light chains: Teaching old dogs new tricks. Bioarchitecture. 4 (6), 169-188 (2014).
- Belyaev, A. S., Hails, R. S., Roy, P. High-level expression of five foreign genes by a single recombinant baculovirus. Gene. 156 (2), 229-233 (1995).
- Li, J., et al. Ca(2+)-induced rigidity change of the myosin VIIa IQ motif-single alpha helix lever arm extension. Structure. 25 (4), 579-591.e4 (2017).
- Persson, M., Bengtsson, E., ten Siethoff, L., Mansson, A. Nonlinear cross-bridge elasticity and post-power-stroke events in fast skeletal muscle actomyosin. Biophys J. 105 (8), 1871-1881 (2013).
Tags

.