Example 1
The enzyme enolase from M. tuberculosis catalyzes the penultimate step of glycolysis and converts 2-phosphoglycerate to phosphoenolpyruvate (PEP), which is an essential intermediate for several metabolic pathways44,45. CryoEM data for the apo-enolase and PEP-bound enolase samples were collected at the same pixel size of 1.07 Å, and image processing was performed with Relion 3.146,47. The apo-enolase and the PEP-enolase structures were determined at 3.1 Å and 3.2 Å, respectively48. The maps and models were deposited in EMDB and PDB49,50 (EMD-30988, EMD-30989, PDB-7e4x, and PDB-7e51). The cryoEM map of the enzyme shows that it is an octamer in the solution (Figure 1A). In order to identify the ligand density in the PEP-enolase map, the unsharpened maps of the apoenzyme and PEP-bound enzyme were selected, and a difference map was calculated in ChimeraX, by subtracting the PEP-enolase map from the apoenzyme map. A distinct density (green) at a high threshold was observed, which suggested the presence of the ligand (Figure 1B). Modeling the protein chain in the unsharpened map clearly indicated that the extra density is present in the active site of the protein (Figure 1C). The ligand, PEP, was then modeled in the B-factor sharpened map using Coot, and the protein+ligand model was refined in real space with Phenix. Two Mg2+ ions were modeled in the density observed in the vicinity of the ligand (Figure 1D). The ligand, PEP, adopts a similar orientation as observed in other enolase homologs, and several active site residues, such as Lys-386, Arg-364, form hydrogen bond interactions with the ligand PEP. The Mg2+ ions form metal coordination bonds with Asp-241, Glu-283, Asp-310, and the phosphate of PEP (Figure 1D).
Example 2
In the absence of an available apo-protein structure or if the protein undergoes a large conformational change, calculating the difference maps as described above is not possible. In 2021, Garib Murshudov's group at the Laboratory of Molecular Biology, Cambridge, introduced Servalcat22, which implements a refinement workflow using Refmac and also calculates a Fo-Fc difference map after refinement. A positive Fo-Fc difference density suggests the presence of molecules/ligands that were not included in the model during refinement, essentially an omit map. However, it is recommended to first assess the fit of the model to the map in general and then evaluate the difference density map.
To illustrate the use of Servalcat/Refmac, mGlu5, a dimeric G-protein coupled receptor that binds to the neurotransmitter, L-Glutamate was chosen. Upon binding of the agonist, L-quisqualate, the extracellular domain reorients, which triggers the rotation of the 7TM, bringing them closer to stabilize the activated state. Thus, there is a large conformational change observed between the apo/antagonist vs. agonist bound states51 (Figure 2A and Figure 2E). The two half-maps for both the agonist (EMD-31536) and the antagonist (EMD-31537) bound complexes were obtained from EMDB, and the cryoEM maps show varied resolution across the molecule and better-resolved extracellular domain. Subsequently, these were utilized as inputs in Servalcat along with the apo-protein as the model to compute the difference or Fo-Fc map for each dataset. This map distinctly showed the presence of various ligand molecules (non-protein). The resolution estimated by FSC (Fourier Shell correlation) for the agonist and antagonist bound complexes were 3.8 Å and 4.0 Å, respectively. In the case of the agonist-bound mGlu5, the Servalcat Fo-Fc difference map showed the presence of both the agonist (L-quisqualate) (Figure 2B) and N-AcetylGlucosamine (NAG) (Figure 2C) in the ECD of the receptor (due to lower resolution of the TMD, here we focus only in the ECD and the top of the TMD). Protein residues, including Tyr-64, Trp-100, Ser-151, and Thr-175, are seen to interact with the agonist. Density near the Asn-210 residue suggested the presence of N-acetyl glucosamine (Figure 2C). A density consistent with cholesteryl hemisuccinate, which was added during the purification of mGlu5, was observed near the top of transmembrane helix 1 (Figure 2D). As the resolution is moderate and the ligand, L-quisqualate, can be placed in different orientations, prior structure of the extracellular domain with the ligand (PDB-6N50) was used as a guide to model the ligand. Antagonist binding stabilizes the open or resting state of the receptor (Figure 2E). A density consistent with the antagonist LY341495 was observed at the hinge of lobe I and lobe II of the venus fly trap domain in the ECD. The antagonist interacts with residues similar to those of the agonist. Stacking interaction between Tyr-223 in lobe II with the antagonist stabilizes the receptor in an open state (Figure 2F). Similar to the agonist structure, glycosylation or the presence of N-acetylglucosamine moiety was observed near Asn-210 (Figure 2G).
Example 3
The third example elucidates the protocol to model fragment-sized or small-sized ligands and solvent molecules in high-resolution CryoEM maps. Fragment-based Drug Discovery (FBDD) has emerged as a powerful and innovative method in the development of target-based novel therapeutics in various disease areas, making it a promising avenue in pharma R&D52,53. FBDD begins with screening and careful selection of small, highly soluble, low-molecular-weight fragments of molecules that bind to specific target proteins or biomolecules of interest. Determining the structures of these protein-fragment complexes reveals the mode of binding of these fragments, which serves as a guide to designing larger and more complex drug-like molecules with increasing affinity and specificity towards the target protein54. However, this method demands a high-resolution ligand density to accurately determine the pose and correctly place the functional groups of the ligand15.
β-galactosidase, one of the first high-resolution structures to be determined from the advancements in cryoEM technology, is a well-studied 450kDa homotetrameric enzyme that catalyzes the hydrolysis of lactose to glucose and galactose55. To showcase the use of cryoEM in FBDD, Astex, UK, determined the structure of β-galactosidase with a fragment-sized inhibitor, deoxygalacto-nojirimycin (DGN) bound in the active site (EMDB-10563, PDB:6tsh)56. This dataset is used to illustrate the protocol to model ligands and solvents unambiguously in high-resolution maps. To showcase the effect of resolution on ligand modeling and visualization, the maps were filtered to 3.0 Å and 3.5 Å in the post-processing step in Relion. This highlights the quality of the map density at different resolutions and underscores the need for higher resolution for modeling ligands and solvents.
The enzyme is a tetramer with D2 symmetry in solution (Figure 3A). Difference map (between map and model), as computed by Servalcat, suggested the presence of DGN and several solvent molecules in the active site of the enzyme (Figure 3B). At an estimated resolution of 2.3 Å, the density showed high-resolution features, which helped in the accurate modeling of the inhibitor in the protein active site. Interactions between DGN and Tyr-503 and His-540 were observed (Figure 3C). The difference density also suggested the presence of solvent molecules that interact with DGN as well as the protein residues. Mg2+ and several water molecules were modeled in the density (Figure 3D). Metal coordination bonds between Mg2+ and Glu-416, Glu-461, and several water molecules are observed (Figure 3D). Mg2+ was seen to interact with DGN via a water molecule.
At a lower resolution of 3.5 Å and 3.0 Å, the ligand density resembles a blob and lacks high-resolution features crucial for accurate modeling of the ligand (Figure 4A,B). Density for water molecules was almost non-existent at these resolutions. In summary, with increasing resolution, especially higher than 3.0 Å, the density enabled the modeling of a greater number of water molecules (Figure 4C,D). The correct positioning of the ligand's chiral centre became achievable at ~2.3 Å (Figure 4C,D) due to the presence of distinct features in the map that guided the placement as well as modeling of water molecules and Mg2+. In comparison, the density for Mg2+ remained discernible across the resolution range (Figure 4A–C).
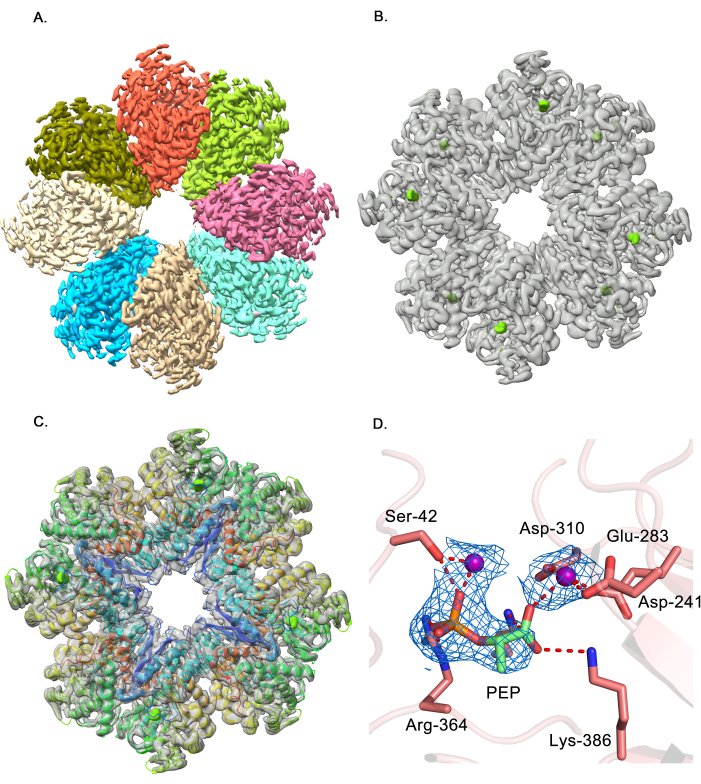
Figure 1: Modeling the ligand phosphoenolpyruvate in M. tuberculosis enolase. (A) shows the B-factor sharpened map of the PEP-bound enolase enzyme. The map suggests that enolase is octameric in solution, and each monomer in the map is colored differently. (B) displays an unsharpened cryoEM map of apo-Enolase enzyme in grey with the difference map (between PEP-bound and apo-Enolase unsharpened maps) overlaid in green, suggesting the presence of ligand, PEP. This is a demonstration map to show the presence of ligands. (C) displays the fit of the enolase model in the unsharpened cryoEM map, highlighting the position of the difference density relative to the protein. The protein model is shown in cartoon representation and colored in Chainbow. This figure shows that the extra density (green) is present in the active site of each monomer. (D) shows the ligand, PEP, encased in the B-factor sharpened map, colored blue. Additionally, density for two Mg2+ ions was also observed, which forms metal coordination bonds with several active site residues, including Ser-42, Asp 241, Glu-283, Asp-310, and ligand atoms. The ligand makes hydrogen bond interactions with Lys-386, Lys-335, and Arg-364. The protein residues are shown in stick representation, and Mg2+ ions are shown as purple spheres. The figures in panels (A–D) were generated with Pymol. Please click here to view a larger version of this figure.
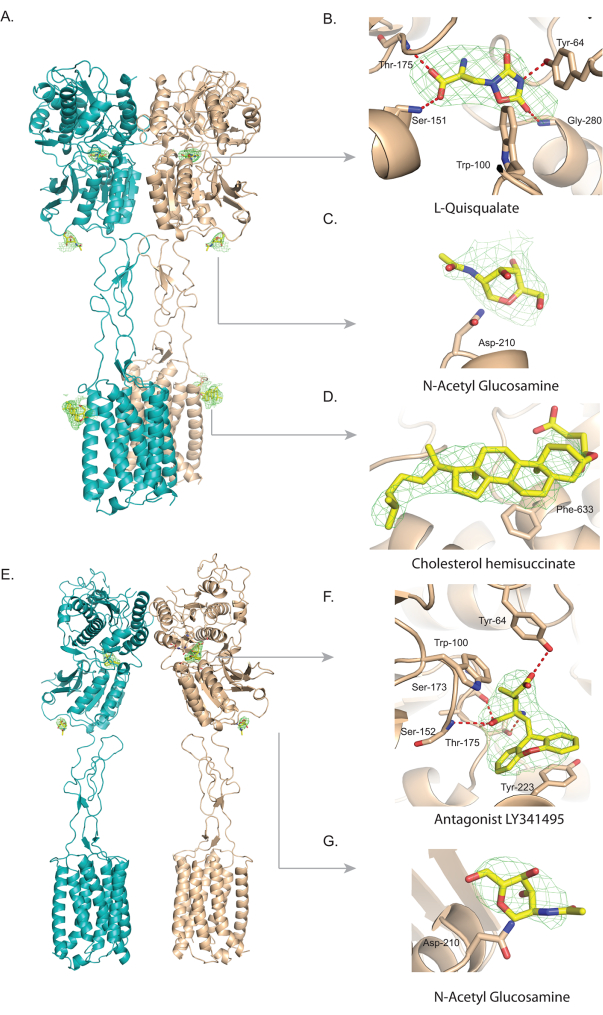
Figure 2: Identification, modeling, and visualization of various ligands in mGlu5 receptor. The Fo-Fc omit maps were obtained using Servalcat. (A) shows mGlu5 receptor dimer structure (PDB-7fd8) in cartoon representation, with each monomer colored in teal and wheat, respectively, and bound to the agonist L-quisqualate. All the ligands that were identified in the extracellular and on the top of the transmembrane domain of the receptor are encased in the Fo-Fc omit map, colored green, and contoured at 6σ.(B) highlights the fit of the agonist, L-quisqualate encased in the difference map. L-quisqualate interacts with several mGlu5 residues including Tyr-64, Trp-100, Ser-151, Thr-175 and Gly-280. The H-bond interactions are represented by red dashes. In (C), an additional density is evident near Asn-210, which is present in the receptor's extracellular domain, and N-acetylglucosamine molecule (NAG) was modeled in this density. For clarity, the NAG is not linked to Asn in the current figure. (D) shows the difference in density for cholesterol hemisuccinate (CHS) in green near the lipid-exposed surface of the receptor. The CHS molecule, represented in the sticks, was modeled in this density. (E) shows mGlu5 receptor structure (PDB-7fd9) bound to the antagonist LY341495 in cartoon representation. In (F), an extra density in the Fo-Fc difference map located at the hinge between lobe I and lobe II of the extracellular domain indicates the presence of the antagonist. Key residues (Tyr-64, Trp-100, Ser-152, Ser-173, Thr-175, and Tyr-223) around the antagonist are shown in stick representations, and potential hydrogen bond interactions with the antagonist are shown in red dashes. (G) shows the difference in density suggestive of the presence of NAG molecule near Asn-210 in the antagonist-bound structure (note, the NAG is not linked with Asn for clarity). The figures were generated with Pymol. Please click here to view a larger version of this figure.
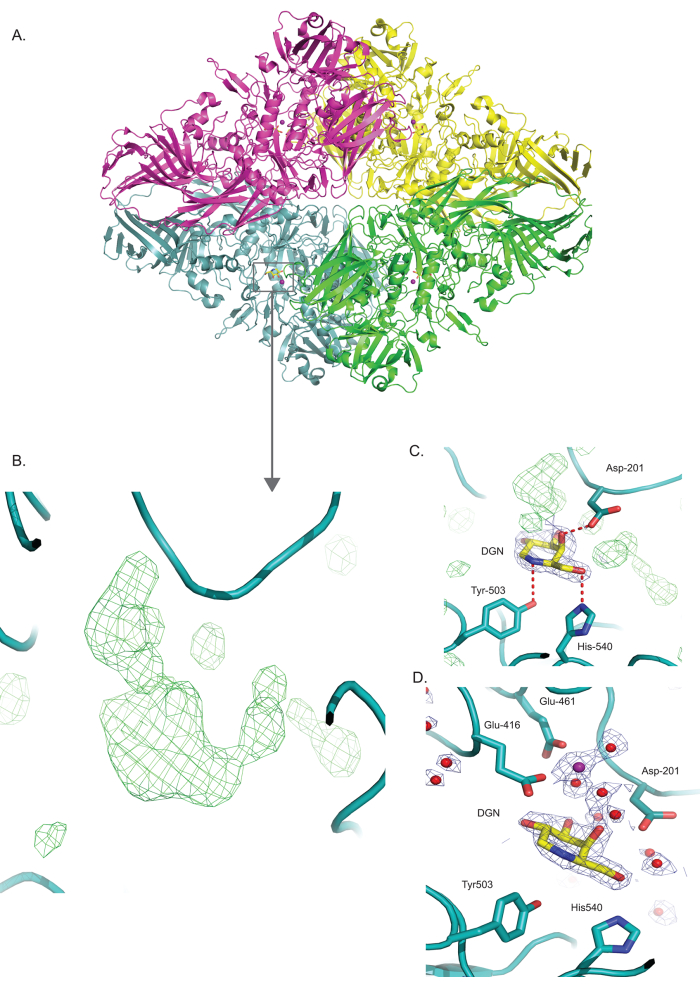
Figure 3: Identification, modeling, and refinement of a small inhibitor and solvent molecules in the high-resolution map of β-galactosidase (EMD-10563). (A) shows the β-galactosidase model resolved to 2.3 Å (PDB: 6tsh) in cartoon representation, where each monomer is colored distinctly. The grey box highlights the ligand binding site. (B) displays the Fo-Fc difference density (from Servalcat) in green mesh in the active site of the enzyme. The difference in density suggests the presence of the inhibitor (DGN) and several solvent molecules in the active site. (C) demonstrates that guided by the Fo-Fc map, the inhibitor deoxygalacto-nojirimycin (DGN) is modeled in the active site. This modeled ligand is depicted in stick format and encased in the Fo density (by Servalcat), which is colored in blue_mesh. Hydrogen bond interactions between DGN and several protein residues, including Tyr-503 and His-540, are observed. The additional Fo-Fc difference density around the ligand (green) is indicative of the solvent molecules. (D) The map shows several solvent molecules, including water and Mg2+, represented as red and purple spheres, respectively, are modeled in the active site after ensuring that each solvent molecule is bonded to either protein (Glu-416, His-418, and Glu-461) or ligand residues. The water molecules and Mg2+ are encased in the Fo density (blue mesh). Mg2+ is seen to interact with the ligand, DGN via a water molecule. The Fo-Fc map (green mesh) in panels (B,C) is contoured at 6σ, whereas the Fo density – blue mesh in C and D (from the Servalcat refinement after modeling) is contoured at 3σ. The figures in panels (A–D) were generated with Pymol. Please click here to view a larger version of this figure.
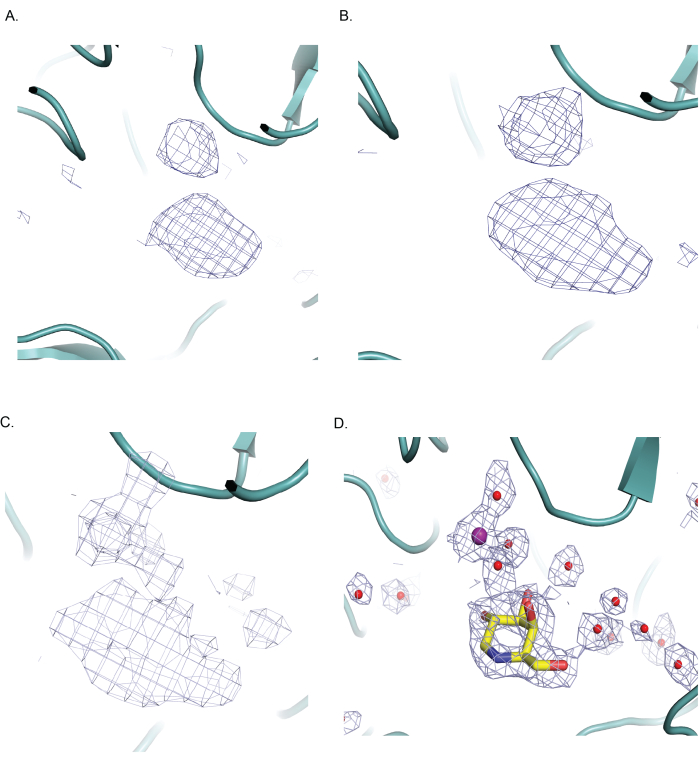
Figure 4: Effect of resolution on ligand modeling in β-galactosidase. Half-maps from EMD-10563 were used as input in the Relion post-process step, and the combined post-process maps were filtered to resolutions 2.3 Å, 3.0 Å, and 3.5 Å with different B-factors. The map shown in all the panels is contoured at 6σ. For clarity, only the protein backbone is shown with no side chains, ligand or solvent molecules in panels A, B, and C. (A) The map is filtered to 3.5 Å, and the active site of β-galactosidase is shown. A blob resembling the ligand, DGN, is seen at this resolution, accompanied by a few smaller blobs in the vicinity. Modeling the ligand in the correct orientation proves challenging due to the lack of distinct features in the map. (B) The map filtered to 3.0 Å resolution is shown. Here, the ligand blob becomes slightly more defined but still lacks features in general. A few more small blobs suggestive of solvent molecules are also observed. (C) The map filtered to 2.3 Å resolution reveals the ligand density with distinct features, notably revealing the chair conformation of the iminosugar. A significant number of small blobs corresponding to water molecules are observed at this resolution. The automatic B-factor estimation/sharpening in Relion post-process gives a value of -18 Å2 for the maps filtered to 3 Å and 3.5 Å, while the value is -52 Å2 for the map filtered to 2.3 Å. Different B-factor sharpening of EM maps can also be performed with Coot and useful in model building. (D) The panel illustrates that the ligand DGN (stick representation), Mg2+, and water molecules (as spheres) as modeled in the active site and the sharpened map at 2.3 Å, shown in blue mesh surrounding these atoms. The figures in panels (A–D) were generated with Pymol. Please click here to view a larger version of this figure.
