The immunofluorescent detection of MLKL phosphorylation and especially RIPK3 phosphorylation in human cells is technically challenging26. We here present an improved staining protocol for human p-RIPK3 (S227) and p-MLKL (S358) upon the activation of ZBP1. The protocol includes a TSA step to improve the detection limit and sensitivity of the fluorescent signals. To validate the method, a side-by-side comparison of the TSA-mediated immunofluorescence with standard indirect fluorescent staining of both p-RIPK3 (S227) and p-MLKL (S358) was performed.
HT-29 cells expressing human ZBP1 were infected for 9 h with an ICP6 RHIM mutant HSV-1 strain (HSV-1 ICP6mutRHIM) to induce ZBP1-mediated necroptosis and RIPK3 phosphorylation. The HSV-1 ICP6mutRHIM strain carries a VQCG to AAAA mutation within the ICP6 RHIM and is unable to block necroptosis signaling downstream of ZBP114,15. As previously reported26, standard indirect immunofluorescence was not sensitive enough to visualize RIPK3 S227 phosphorylation with the current commercially available antibody, even when the laser power of the confocal microscope was increased to 30% (Figure 2A). In contrast, the inclusion of a TSA step enabled the robust detection of p-RIPK3 (S227) in the cytosol of cells infected with HSV-1 ICP6mutRHIM. The p-RIPK3 (S227) signal reached saturation when the laser power was set at 2% (Figure 2A). Quantification of the three-dimensional z-stack images (see step 9) showed an approximate 20-fold increase in the number of voxels that were positive for p-RIPK3 (S227) in HSV-1 ICP6mutRHIM-infected over mock-treated cells (Figure 2B). Omitting the primary anti-p-RIPK3 (S227) antibody from the TSA-mediated staining protocol as a no primary (NP) control did not yield a detectable signal. To visualize HSV-1 ICP6mutRHIM-infected cells, the samples were co-stained with a primary antibody directed against the immediate early viral protein ICP0 (Figure 2A). A low but detectable p-RIPK3 (S227) signal was present in the mock-treated cells, which may represent the constitutive autophosphorylation of human RIPK3 at this site5 (see the discussion). In line with a previous report28, the RHIM of ICP6 is unable to fully block RIPK3 S227 phosphorylation, as we detected increased p-RIPK3 (S227) staining of cells infected with wild-type HSV-1 (HSV-1WT; Figure 2A,B). To further validate the specificity of the p-RIPK3 (S227) signal, the cells were treated with the RIPK3 kinase inhibitor GSK'840 prior to infection. GSK'840 binds to the kinase domain of RIPK3, preventing its activity and thereby inhibiting its autophosphorylation29. GSK'840 prevented RIPK3 phosphorylation at S227 upon ZBP1 activation (Figure 2A,B), confirming the specificity of the TSA-mediated p-RIPK3 (S227) detection method.
To follow MLKL phosphorylation, an end-stage marker for necroptosis, ZBP1-expressing HT-29 cells were infected with HSV-1 ICP6mutRHIM for 8 h and 10 h. The cells were stained with an antibody against S358 phosphorylated MLKL (p-MLKL [S358]) using TSA. The mock-treated cells showed low and somewhat punctate cytosolic staining of p-MLKL (S358), while strong p-MLKL staining was detected in the cytosol, nucleus. and at the plasma membrane in the cells that were infected with HSV-1 ICP6mutRHIM (Figure 3A). Moreover, the p-MLKL (S358) signal was observed in clusters. This is in line with the pore-forming functions of activated phosphorylated MLKL oligomers at the cell membrane and its recently reported nuclear translocation upon influenza A infection1,2,6,30. As a positive p-MLKL (S358) staining control, we stimulated ZBP1-expressing HT-29 cells with a combination of TNF, the SMAC mimetic BV6, and pan-caspase inhibitor zVAD-fmk to induce TNFR1-mediated necroptosis (Figure 3A). Omitting the primary anti-p-MLKL (S358) antibody from the TSA-mediated staining protocol as a no primary control did not yield a detectable signal.
Next, we performed a side-by-side comparison of p-MLKL (S358) immunofluorescent staining with and without TSA. ZBP1-expressing HT-29 cells were infected for 9 h with HSV-1 ICP6mutRHIM. While a laser power of 40% was needed to detect a specific p-MLKL (S358) signal in the infected cells using standard indirect immunofluorescence, the TSA-treated samples already reached saturating signals at a laser power of 6% without increasing the background staining in the mock-treated samples (Figure 3B). Moreover, quantification of the three-dimensional z-stack images showed an over 10-fold increase in the number of voxels that were positive for p-MLKL (S358) when using TSA compared to standard indirect immunofluorescence. This shows that TSA improves both the detection threshold and sensitivity for p-MLKL (S358; Figure 3C).
Finally, to validate the TSA-mediated immunofluorescence protocol for other ZBP1-dependent necroptosis viral stimuli, we infected ZBP1-expressing HT-29 cells with influenza A virus (IAV) PR8 strain for 9 h. IAV induces both ZBP1-mediated apoptosis and necroptosis in human cells30. Indeed, TSA allowed the robust detection of p-RIPK3 (S227) and p-MLKL (S358), indicative of these cells undergoing necroptosis (Figure 4A–D).
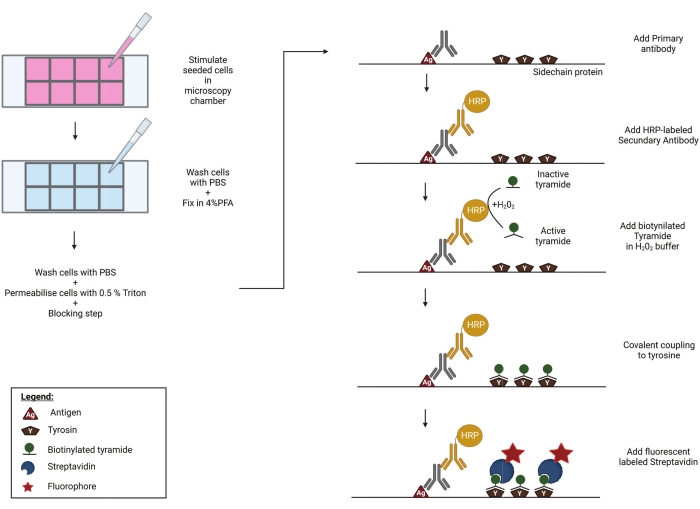
Figure 1: Schematic representation of the TSA protocol. Cells are seeded and stimulated in a well plate, compatible with high-end microscopy. Afterward, the samples are fixed in 4% PFA, permeabilised and blocked to prevent aspecific binding of primary antibodies. In order to visualize phosphorylated RIPK3 (p-RIPK3 [S227]) and MLKL (p-MLKL [S358]), specific antibodies recognizing these key phosphorylation sites are incubated overnight on the imaging chamber. Next, a secondary antibody, coupled to a horse radish peroxidase (HRP), is added. This HRP group enables the activation of biotinylated tyramide in the presence of H2O2. Subsequently, the active biotin-tyramide covalently couples to tyrosine residues in close proximity to the HRP-labeled secondary antibody. These include tyrosines on the proteins of interest-in this case p-RIPK3 or p-MLKL, as indicated in the figure-and those of neighboring proteins and on the primary and secondary antibodies themselves (not shown). This tyramide signal amplification step greatly increases the sensitivity of the staining protocol. In a final step, streptavidin coupled to a fluorescent group is added to visualize the biotinylated molecules. Please click here to view a larger version of this figure.
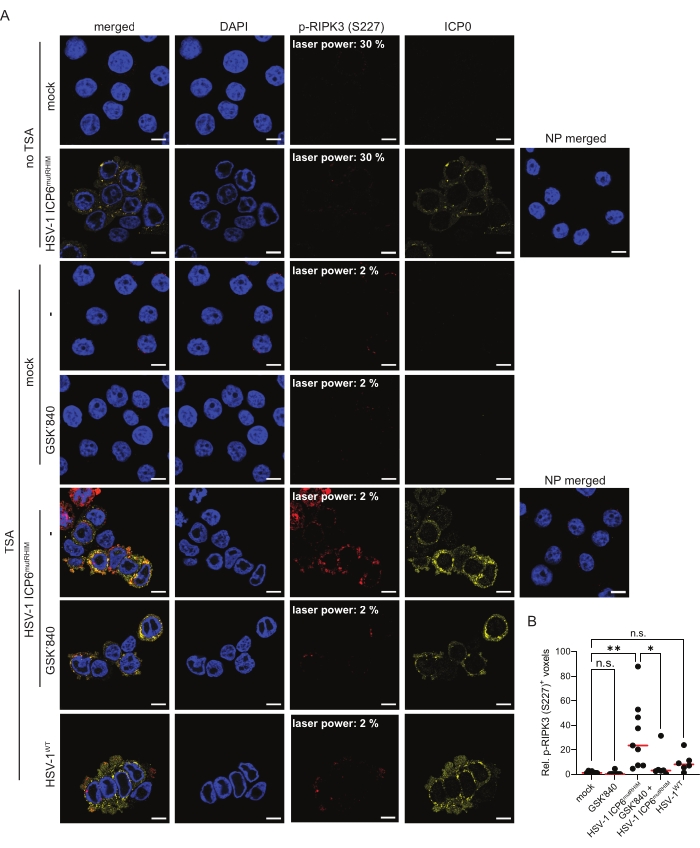
Figure 2: HSV-1 ICP6mutRHIM induces ZBP1-dependent phosphorylation of human RIPK3 at S227. (A) Representative confocal images of human ZBP1-expressing HT-29 cells, comparing TSA staining with a standard indirect (no TSA) immunofluorescence staining protocol for p-RIPK3 (S227). The mock- and virus-infected samples (HSV-1WT or HSV-1 ICP6mutRHIM [MOI = 5]) were incubated for 9 h. As a negative control, the RIPK3 kinase inhibitor GSK'840 (1 µM) was included. A no primary (NP) staining control of cells infected with HSV-1 ICP6mutRHIM (MOI = 5) for 9 h in which both the primary anti-p-RIPK3 (S227) and ICP0 antibodies were omitted is included. The laser power necessary to detect the specific p-RIPK3 (S227) signal is indicated on the images. ICP0 was used to stain the virus-infected cells, and DAPI was used to stain the nucleus. The scale bars are 10 µm. (B) Relative quantification of p-RIPK3 (S227)+ voxels using the TSA staining protocol. Every dot represents an image, and the red bar represents the median. Voxel count values are presented relative to the median of the voxel count of images in the mock condition. Statistics were done using a one-way ANOVA with multiple comparisons using Tukey correction. p > 0.05 (n.s.), p≤ 0.05 (*), p≤ 0.01 (**). Please click here to view a larger version of this figure.
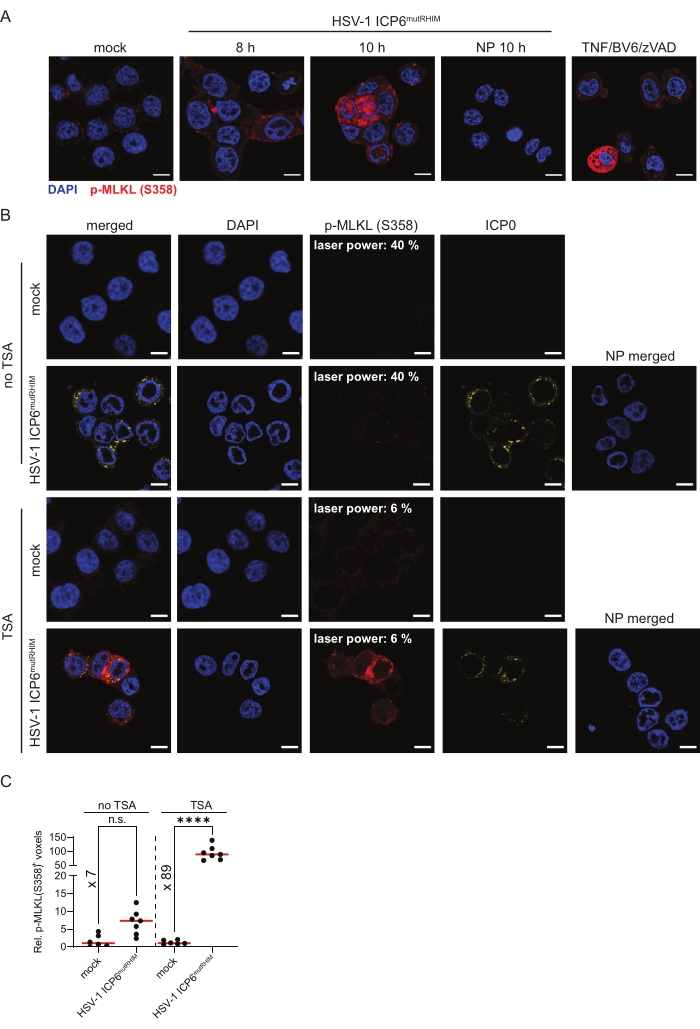
Figure 3: HSV-1 ICP6mutRHIM induces ZBP1-dependent phosphorylation of human MLKL at S358. (A) Representative confocal images of human ZBP1-expressing HT-29 cells. The cells were either mock treated or infected with HSV-1 ICP6mutRHIM (MOI = 5) for 8 h and 10 h. TSA was used to detect p-MLKL (S358). ICP0 was used to stain the virus-infected cells, and DAPI was used to stain the nucleus. A no primary (NP) staining control of cells that were infected for 10 h with HSV-1 ICP6mutRHIM (MOI = 5) in which the primary anti-p-MLKL (S358) was omitted is shown. As a positive control, the cells were stimulated for 4 h with 30 ng/mL TNF, 5 µM BV6, and 20 µM ZVAD-fmk, which induces necroptosis via TNFR1. The scale bars are 10 µm. (B) Representative confocal images of human ZBP1-expressing HT-29 cells, comparing TSA staining with a standard indirect (no TSA) immunofluorescence staining protocol for p-MLKL (S358). The cells were either mock treated or infected with HSV-1 ICP6mutRHIM (MOI = 5) for 9 h. An NP staining control of cells infected with HSV-1 ICP6mutRHIM (MOI = 5) for 9 h in which the primary anti-p-MLKL (S358) and ICP0 antibodies were omitted is included. The laser power necessary to detect the specific p-MLKL (S358) signal is indicated on the images. (C) Relative quantification of p-MLKL (S358)+ voxels using the standard (no TSA) and TSA staining protocol. Every dot represents an image, and the red bar represents the median. The voxel count values are presented relative to the median of the voxel count of images in the mock condition. Statistics were done using a one-way ANOVA with multiple comparisons using Tukey correction. p > 0.05 (n.s.), p ≤ 0.0001 (****). Please click here to view a larger version of this figure.
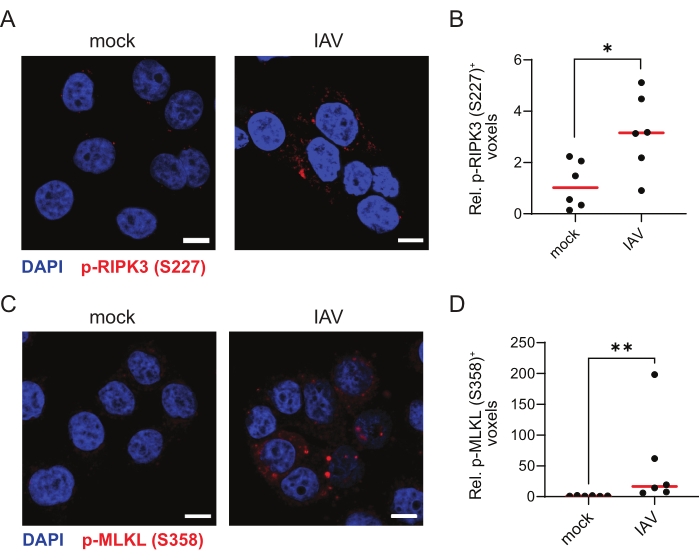
Figure 4: Influenza A virus induces ZBP1-dependent phosphorylation of human RIPK3 and MLKL. (A,C) Representative confocal images of human ZBP1-expressing HT-29 cells. The cells were either mock treated or infected with influenza A virus (IAV), PR8 strain (MOI = 4) for 9 h and stained for p-RIPK3 (S227; A) or p-MLKL (S358; C) using the TSA protocol. The scale bars are 10 µm. (B,D) Relative quantification of p-RIPK3 (S227)+ (B) or p-MLKL (S358; D) voxels. Every dot in (B,D) represents an image, and the red bar represents the median. The voxel count values are presented relative to the median of the voxel count of images in the mock condition. Statistics were done using a Mann-Whitney test. p≤ 0.05 (*), p ≤ 0.01 (**). Please click here to view a larger version of this figure.
