1. Preparation of buffers
NOTE: Use ultrapure H2O for all buffers. It does not have to be sterile. Filter all the solutions described in steps 1.1-1.4 with a 0.2 µm syringe filter, aliquot in portions of 500 µL-2 mL per tube depending on usage, and store at -20 °C.
- Prepare 10% BSA by weighing 2 g of BSA into a 50 mL conical tube and filling to the 20 mL mark with H2O. Mix until the BSA is dissolved (about 30 min), and then make up the volume to 20 mL.
NOTE: Use high quality BSA (see Table of Materials). The 10% BSA solution is used in both bead preparation and the motility mix. - For bead preparation, prepare Xb buffer (10 mM HEPES, 0.1 M KCl, 1 mM MgCl2, and 0.1 mM CaCl2, pH 7.5) as a 10x solution, and dilute before use (100 µL of 10x Xb stock solution + 900 µL of H2O). Prepare Xb/1% BSA by mixing 100 µL of 10x Xb stock solution + 100 µL of 10% BSA + 800 µL of H2O.
- Prepare G-buffer (2 mM Tris, 0.2 mM CaCl2, 0.2 mM DTT, 2 mM ATP), which is the buffer used to dilute monomeric actin (G-actin). Adjust to pH 7, not pH 8 as traditionally used (see discussion).
- Prepare the motility buffer MB13 (10 mM HEPES, 1.5 mM ATP, 3 mM DTT, 1.5 mM MgCl2, 1 mM EGTA, 50 mM KCl, 1% BSA, pH 7.5). For some applications, 10x MB13 is useful. However, prepare 10x MB13 without BSA, as it leads to problems during pH adjustment. Add BSA from 10% stock solution (prepared in step 1.1) when reconstituting 1x MB13 from 10x MB13.
2. Preparation of protein solutions
NOTE: Use ultrapure H2O for all resuspensions. It does not have to be sterile. Handle all proteins on ice and aliquot into pre-chilled tubes. Manipulate gently so as not to produce bubbles, and never vortex protein solutions. For stocks to be stored at -80°C, flash freezing in liquid nitrogen is not necessary. Adapt aliquot size to avoid more than about five freeze-thaw cycles, as this does not appear to affect the activity of any of the proteins. Working aliquots can be stored at -20 °C for a few weeks.
- Prepare G-actin (rabbit skeletal muscle) solution as described below.
- Pulse centrifuge actin powder (see Table of Materials) at 4 °C to gather the solid at the bottom of the tube.
- Add H2O as per manufacturers' instructions (1 mg protein in 100 µL of cold H2O for unlabeled actin, 100 µg protein in 100 µL of cold H2O for ATTO-labeled actin).
- Let it sit on ice for at least 15 min. Mix gently by pipetting up and down, let it sit on ice at least another 15 min, and mix again. Pulse centrifuge at 4 °C to collect the solution at the bottom of the tube, and remix.
- Prepare 10-50 µL aliquots of unlabeled actin depending on use, and 20 µL aliquots of ATTO-labeled actin. Store the aliquots at -80 °C.
- To depolymerize actin oligomers that form during lyophilization and freezing, dilute an aliquot of resuspended actin ~8-fold in G-buffer spiked with additional ATP and DTT (for example, to 20µL of resuspended actin solution from step 2.1.4, add 134 µL of G-buffer, 0.32 µL of 0.2 mM ATP and 0.16 µL of 1 M DTT). For fluorescent labeling, add approximately 10% labeled actin; for example, add 5 µL of ATTO-labeled actin to 40 µL of diluted unlabeled actin. Let it depolymerize on ice with occasional mixing (pipetting) at least a few days to a week before measuring protein concentration by Bradford assay as described in step 3.
NOTE: Keep diluted unlabeled and fluorescent actin on ice in a cold room or refrigerator; never freeze or allow to warm. The preparation will continue depolymerizing over time, and when handled properly can be used for at least 6 months.
- Resuspend Arp2/3 complex (porcine brain) (see Table of Materials) following the manufacturer's instructions (20 µg protein in 20 µL cold H2O), with the sequence of pulse centrifuging, mixing, etc. on ice as described for actin in step 2.1. Combine the protein solutions from resuspending two tubes of powder to have a larger stock for reproducible experiments. Prepare 2 µL aliquots and store at -80 °C.
- Resuspend profilin (human recombinant) (see Table of Materials) at a 4x higher concentration than stipulated in the manufacturer's instructions (100 µg protein in 25 µL cold H2O), with the sequence of pulse centrifuging, mixing, etc. on ice as for actin in step 2.1. Combine the protein solutions from resuspending two tubes of powder before determining protein concentration to have a larger stock for reproducible experiments.
NOTE: Keep on ice in a cold room or refrigerator; never freeze or allow to warm. When handled properly, resuspended profilin is good for at least 6 months to 1 year. - Resuspend capping protein (α1β2, human recombinant) (see Table of Materials) following the manufacturer's instructions (50 µg protein in 50 µL cold H2O), with the sequence of pulse centrifuging, mixing etc. on ice as described for actin in step 2.1. Chill 50 µL of glycerol on ice and add the 50 µL of resuspended capping protein to it; mix gently. Store at -20 °C.
NOTE: The solution does not freeze and the activity is robust, so the solution can be kept as a single aliquot for months, or even years, if handled carefully. Mouse recombinant capping protein, the one most commonly used in the past in in vitro experiments13, will soon be commercially available. - Resuspend gelsolin (human recombinant, His-tagged) (see Table of Materials) following the manufacturer's instructions (20 µg protein in 20 µL cold H2O), with the sequence of pulse centrifuging, mixing, etc. on ice as described for actin in step 2.1. About 2 µL of gelsolin is used per day of experiments; therefore, prepare large aliquots (5-10 µL) and store at -80 °C.
NOTE: The protocol with gelsolin is provided as an alternative. The use of capping protein in the place of gelsolin is recommended, either purchased or purified as in13. - Resuspend VCA (human WASP-VCA, GST-tagged) (see Table of Materials) at 2x higher concentration than stipulated in the manufacturer's instructions (500 µg protein in 250 µL cold H2O), with the sequence of pulse centrifuging, mixing, etc. on ice as for actin in step 2.1. Make 10 µL aliquots and store at -80 °C.
NOTE: Once SpVCA (human pVCA, streptavidin and His-tagged) is commercialized or if protein purification14 is possible, the use of SpVCA instead of VCA is recommended. VCA does not reproducibly give comets under the conditions described here.
3. Measurement of protein concentrations
- Construct a Bradford standard curve made of two overlapping serial dilutions of BSA.
NOTE: The standard curve only needs to be constructed every couple of months (or even less frequently) as long as the spectrophotometer does not change.- In row 1 of a microtube rack, place tubes for BSA dilution series #1: four 2 mL tubes followed by four 1.5 mL tubes. In row 3 of the rack, place tubes for BSA dilution series #2 as for series #1. In row 5 of the rack, place one 2 mL tube for each sample to be measured along with two 1.5 mL tubes. Add a single 1.5 mL tube in row 5 for the blank.
- Measure out Bradford Reagent (see Table of Materials) into the 1.5 mL tubes.
- Fill a 15 mL conical tube to the top with Bradford Reagent for easier pipetting (excess will be returned to the stock bottle in the refrigerator) on ice. Take up 200 µL Bradford Reagent and eject it back into the conical tube to wet the pipette tip. As the solution is viscous, pipette slowly to allow the solution to enter and leave the tip completely without making bubbles.
- With the "pre-wet" tip, slowly pipette 200 µL of Bradford Reagent into each of the 1.5 mL tubes in the rack (four for each BSA dilution, one for the blank and two for each sample to be measured). Do this first to allow the Bradford Reagent to thoroughly warm to room temperature before mixing with protein solutions. Return the remaining contents of the 15 mL conical tube to the bottle.
- Measure out H2O into the 2 mL tubes. In row 1 of the rack, add 1,990 µL H2O to the first tube, and 900 µL to the three other tubes. For row 3, add 1,992.5 µL to the first tube and 900 µL to the three other tubes. Add 2,000 µL H2O to each of the sample tubes in row 5.
NOTE: For all volumes larger than 1,000 µL, use a 1,000 µL pipette, but administer the full amount by pipetting twice. It is important to get everything ready before starting the subsequent steps, in order to avoid leaving proteins highly diluted in H2O for long periods of time. - To prepare BSA dilution series #1, mix 10 µL of calibrated 2 mg/mL BSA (see Table of Materials) into the tube with 1,990 µL H2O to make a 10 µg/mL solution. From this, make three serial dilutions (5 µg/mL, 2.5 µg/mL, and 1.25 µg/mL BSA) by transferring 900 µL of each solution into the next tube (containing 900 µL H2O).
- To prepare BSA dilution series #2, mix 7.5 µL of calibrated 2 mg/mL BSA into the tube with 1,992.5 µL H2O to make a 7.5 µg/mL solution. From this, make three serial dilutions (3.75 µg/mL, 1.875 µg/mL and 0.9375 µg/mL BSA) by transferring 900 µL of each solution into the next tube (containing 900 µL H2O).
- Mix and read absorbance to generate the standard curve. Add 800 µL H2O to the Bradford reagent tube for the blank, and start the timer. As efficiently as possible without making bubbles, mix 800 µL of each BSA standard with 200 µL Bradford reagent in the prepared tubes. Once all the standards have been mixed with Bradford reagent (< 5 min), pour each standard into a disposable cuvette and read the absorbance at 600 nm in the spectrophotometer after blanking the machine.
NOTE: The same cuvette can be used to read the whole series if the least concentrated standard is read first and the cuvette is well-emptied between reads. Redo the standard curve until the linear fit has an R value of at least 0.99. Only after pipetting and gentle mixing is mastered, proceed to reading samples.
- Measure concentrations of actin and actin-binding proteins
- In the 2 mL tubes containing 2,000 µL of H2O (prepared in step 3.1.3), gently mix in the following: 2 µL each of Arp2/3 complex and profilin, 4 µL of labeled G-actin, 5 µL each of gelsolin and VCA and 8 µL of capping protein (human recombinant). Immediately take 800 µL of the solution and mix with the already prepared Bradford reagent, and then repeat to have two readings within 5%-10 % of each other for each sample. A larger difference indicates a problem with resuspension or handling. Read within a few minutes of mixing with Bradford reagent.
NOTE: If using a standard curve from another day, only the blank from step 3.1.6 has to be prepared, in addition to the samples.
- In the 2 mL tubes containing 2,000 µL of H2O (prepared in step 3.1.3), gently mix in the following: 2 µL each of Arp2/3 complex and profilin, 4 µL of labeled G-actin, 5 µL each of gelsolin and VCA and 8 µL of capping protein (human recombinant). Immediately take 800 µL of the solution and mix with the already prepared Bradford reagent, and then repeat to have two readings within 5%-10 % of each other for each sample. A larger difference indicates a problem with resuspension or handling. Read within a few minutes of mixing with Bradford reagent.
- Calculate the concentrations of actin and actin-binding proteins using the standard curve and the dilution factor. Redo the measurement for each new resuspension. Molecular weights to convert mg/mL readings obtained via Bradford assay to µM are: actin 43 kD, Arp2/3 complex 224 kD, profilin 15 kD, gelsolin 95 kD, capping protein 68 kD (human recombinant) or 63.5 kD (mouse recombinant), VCA 43 kD, and SpVCA 54 kD (monomer molecular weight).
4. Coating of beads
- Pre-chill the centrifuge to 4 °C and set the agitating dry block (see Table of Materials) to 18 °C.
- Wash the beads: pipette 50 µL of Xb buffer into a 1.5 mL microcentrifuge tube, add 9 µL of 4.5 µm diameter bead suspension or 2 µL of 1 µm diameter bead suspension (2.5 % w/v suspension) (see Table of Materials). Mix thoroughly and centrifuge the samples at 20,000 x g for 10 min at 4 °C.
NOTE: Total bead surface area of both bead sizes is 3 cm2:
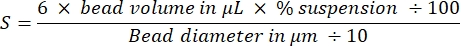
derived by calculating the number of beads and then their total surface using classic equations for the volume and surface area of spheres. Other sizes of beads can be used if amounts are adjusted to keep the total surface area constant at 3 cm2. - Coat the beads: carefully remove the supernatant without disturbing the beads, and resuspend the bead pellet in 40 µL of 2 µM SpVCA (or 7 µM VCA) in Xb buffer by gentle pipetting. Agitate at 18 °C, 1,000 rpm for 20 min.
- Wash coated beads: centrifuge the mix (20,000 x g for 10 min at 4 °C) and carefully remove the supernatant. Resuspend the beads in 50 µL of cold Xb/1% BSA, and centrifuge at 20,000 x g for 10 min at 4 °C. Remove the supernatant and repeat the washing step 1x.
- Resuspend the coated bead pellet from step 4.4 in 120 µL cold Xb/1% BSA for both sizes of beads so that the amount of bead surface area/µL of bead solution is the same. Store on ice in a refrigerator or cold room. Coated beads will continue to function normally for at least several weeks.
5. Preparing the motility mix and slides for observation
NOTE: The total volume of the motility mix is 8.4 µL to allow for about 25 µm clearance between the slide and the 18 mm x 18 mm coverslip so beads of all sizes (up to 10 µm diameter) are not squeezed. The basic motility mix is approximately 5 µM G-actin (10% labeled fluorescent actin) with 5 µM profilin, 50 nM Arp2/3 complex, and 25 nM capping protein (or 240 nM gelsolin).
- Prepare the motility reaction mixture. Exact amounts of actin (and therefore profilin) depend on the concentration calculated in step 3.3, but a representative reaction is as follows. Mix on ice, in this order: 3.2 µL of MB13, 1.5 µL of profilin at 30 µM diluted in MB13, 1 µL of capping protein at 0.21 µM diluted in MB13 or gelsolin diluted to 2 µM in MB13, 1 µL of Arp2/3 complex at 0.47 µM diluted in MB13, 0.2 µL of bead suspension (vortex just before use), and 1.5 µL of actin at 30 µM in G-buffer. Mix well but quickly and start the timer.
- Spot the entire motility reaction mixture on a slide. Cover with an 18 mm x 18 mm coverslip and seal the coverslip with melted VALAP using a small paintbrush. VALAP is a mix of lanolin, paraffin, and petroleum jelly (see Table of Materials) 1:1:1 by weight, melted and stirred together.
6. Microscopy observation
- Observe motility reactions immediately using a 100x objective on either an upright or an inverted microscope (see Table of Materials), equipped with phase contrast and/or epifluorescence microscopy (GFP cube, see Table of Materials). Observations are done at room temperature (23-25 °C).
- To obtain average displacement speeds for a whole population of beads, record phase contrast or fluorescent still images over time by scanning the entire slide. Measure comet length by hand and plot versus time. The slope of the linear fit is the average growth speed.
- To evaluate the speed of individual beads, collect time-lapse movies in phase contrast microscopy. Depending on the bead speed and the resolution required, take frames every 1-10 s. Use the tracking tool of any image processing program to obtain bead speeds and trajectories.
One of the key aspects of reproducibly creating actin comets on beads is gentle and precise pipetting of delicate actin-binding proteins. Generating a Bradford standard curve is a good way of evaluating pipetting skills. Figure 1A,B shows the tubes for the standard curve and an example of what the two serial dilutions of BSA look like once mixed with Bradford reagent. Note the graded blue hue (higher protein concentration gives a bluer solution). When read in the spectrophotometer and plotted, these solutions give a standard curve as shown in Figure 1C. To practice careful pipetting, the assay should be repeated until the linear correlation factor is 0.999, as shown.
Once the concentrations of commercial resuspended proteins have been carefully evaluated via the Bradford assay, coated beads and motility mix are prepared and mixed together. Figure 2A shows representative images of the different stages of comet formation: actin clouds form within minutes of mixing SpVCA-coated beads and motility medium; cloud polarization occurs at ~5 min and comet production at 15-20 min. The actin comets, visible with both epifluorescence and phase contrast microscopy (Figure 2A), continue to elongate for many hours, but a consistent speed is not maintained so bead motility is normally evaluated within 1 h. On the other hand, it takes 30 min with VCA-coated beads to obtain bright actin clouds (Figure 2B), and no comets form, although symmetry starts to break at 1-2 h (arrow in Figure 2B) and clouds show polarization after overnight incubation.
Figure 3 shows an example of bead velocity evaluation in the presence of capping protein. Since all beads break symmetry at approximately the same time, "pseudo-time-lapse" recordings are performed where the slide is scanned and pictures of the whole population of comets are taken over time (Figure 3A). Comets do not depolymerize; therefore, the increase in comet lengths measured over time can be used to calculate displacement speed (Figure 3B). Gelsolin can be used in the place of capping protein for comet formation if 10x more gelsolin is added to compensate for its reduced capping activity. Comets formed in the presence of gelsolin are qualitatively the same and move at approximately the same speeds as beads with capping protein (Figure 3C). Capping activity is key for concentrating polymerization at the surface of the bead, and when neither capping protein nor gelsolin are included in the motility mix, actin clouds never polarize to form comets, although bright actin clouds form around the beads (Figure 3D). Comets on beads can be used to measure actin-based force production in different biochemical contexts by altering the motility mix and observing the result on motility using different micromanipulation techniques, for example15.
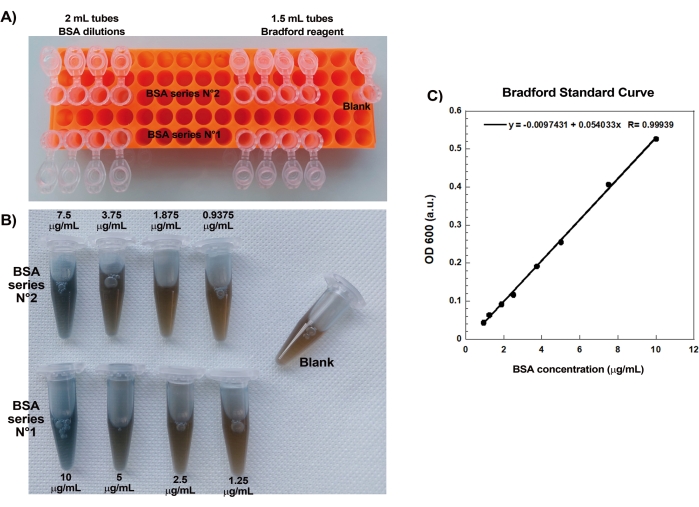
Figure 1: Bradford standard curve. (A) Picture of how to set up the tubes for making the Bradford standard curve. Sample tubes are not shown. (B) Picture of the two overlapping BSA serial dilutions once mixed with Bradford reagent. (C) The absorbances at 600 nm of the solutions shown in (B) are measured in the spectrophotometer and are plotted as a function of the protein concentrations of the BSA solutions. The linear fit is used to calculate the sample concentration. The correlation factor, R, of the linear fit is 0.999. Please click here to view a larger version of this figure.
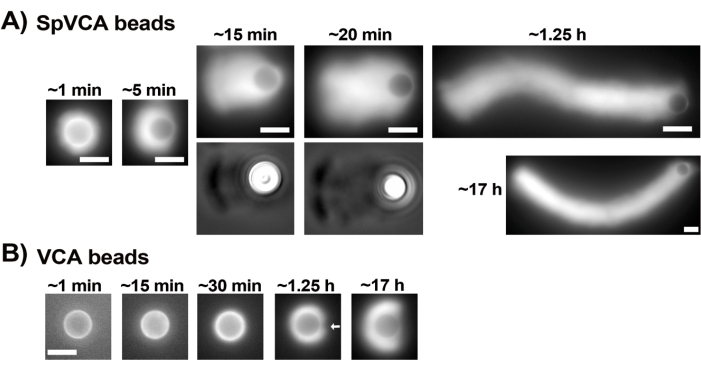
Figure 2: Comet formation on SpVCA-coated beads as opposed to VCA-coated beads. (A) Representative SpVCA-coated beads (different bead in each image) shown over time. Time from the moment of mixing is indicated. Actin clouds form immediately, and cloud polarization gives comets, which continue to elongate for hours. (B) Representative VCA-coated beads (different bead in each image) shown over time. More than 1 h is necessary to see the beginnings of actin cloud polarization (arrow) and even long incubations do not produce comets. All images are of 4.5 µm diameter beads, epifluorescence imaging of fluorescent actin paired with phase contrast visualization for the 15 and 20 min time-points in (A), scale bars = 5 µm. Please click here to view a larger version of this figure.
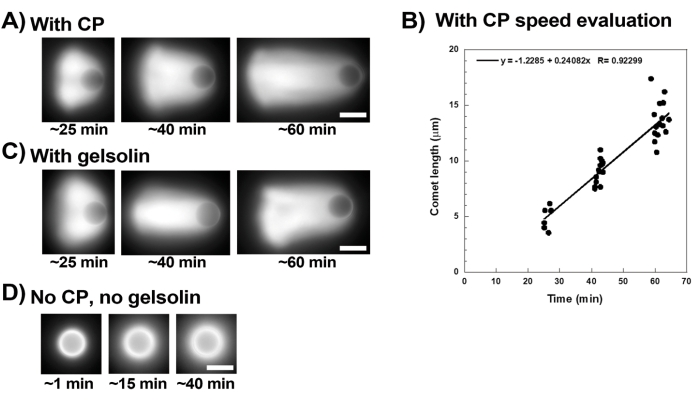
Figure 3: Comets and bead velocity analysis. (A) and (C) In the presence of either capping protein (CP) or gelsolin, actin clouds polarize to form comets in the first 20 min of reaction, and comets elongate over time. Time from mixing indicated for each image; each image is a different bead. There is some variability among beads and preparation, but on average beads move at micron/sub-micron per min speeds (0.2-1 µm/min) under the standard conditions described here. (B) Representative graph showing evaluation of comet length (whole population of beads) over time. The slope of the linear correlation corresponds to the average displacement speed, in this case 0.24 µm/min. (D) In the absence of capping activity (no capping protein or gelsolin), actin clouds form around beads, but comets do not form. All images are of 4.5 µm diameter beads, epifluorescence imaging of fluorescent actin, scale bars = 5 µm. Please click here to view a larger version of this figure.
