After establishing the working principle of the imaging and analysis method, image registration was done to evaluate the expansion ratio and to ensure isotropic expansion during sample processing (Figure 1A,B). Both untreated and VISTA samples were imaged while targeting the bond vibration at 2940 cm−1, which originates from CH3 of endogenous proteins. In untreated samples, the protein-rich structures like nuclei were dark due to the overwhelming lipid content from surrounding tissues22 (Figure 1A). After sample processing that includes the delipidation treatment, the resulting image showed the same feature with an inversed contrast (Figure 1B). The shapes and the relative positions of nuclei and vessels were completely unaltered (Figure 1A,B; numbered structures), confirming that the treatment is an isotropic process. By comparing the sizes of the corresponding nuclei, it was concluded that the method achieves 3.4 times expansion in brain tissue samples as compared to untreated samples22,23.
Knowing the expansion ratio in brain tissue, VISTA can now resolve new features in the label-free SRS images that were previously unresolvable. Although actin and tubulin structures have been the gold standard for super-resolution demonstrations, the resolution improvements on actin and tubulin structures have been well-characterized by fluorescence-based sample-expansion strategies using similar hybridization chemistry28. Moreover, imaging specific actin/tubulin structures is less feasible with this technique because the signal comes from the total ensemble of endogenous proteins, where cytoskeleton structures like tubulins would not have sufficient contrast (signal-to-background ratio) to be clearly distinguished. Hence, we decided to pursue imaging other nanoscale structures. We showed that features can be captured from mouse cortexes down to 150 nm (Figure 1C,D). Based on the dispersive patterns around neuronal dendrites, the observed small structures are likely dendritic spine heads7, which have a size of 146 nm (Figure 1D). In addition, the method was used to image the fibrillar structures in Aβ plaques, which are believed to have a thickness around 100 nm29,30. Indeed, it was demonstrated that ~130 nm fibrillar structures can be resolved in a representative diffusive Aβ plaque using this method (Figure 1E,F).
As VISTA enables effective protein retention and protein imaging22, one can clearly distinguish protein-rich nucleoli in nuclei and the ribbon-like cytoskeleton structures in the cytosols of cultured HeLa cells (Figure 2A, arrowhead). The method was further applied to study poly-glutamine (polyQ) aggregates that are transiently expressed in mammalian cells (Figure 2B,C). The results confirmed that the aggregates, as an expectedly densely packed structure, were expanded isotropically by comparing the same aggregate structures before and after expansion across multiple replicate samples23. High-resolution structures that are absent/blurred in the normal-resolution SRS images were obtained using this method. The VISTA-aggregate images revealed fibril-like protrusions on the periphery of the polyQ aggregates and a hollow structure in the center (Figure 2B, arrowhead). The observation that protrusions seamlessly attach to cytosolic contents might suggest that aggregates engage with functional proteins in cytosol. In hindsight, the capacity to expand dense aggregates also becomes plausible because the fixation reagent formaldehyde and the hydrogel monomers acryl amide and sodium acrylates are all small molecules that can diffuse in and out of protein aggregates. Once the aggregate co-polymerizes with monomers into hydrogel, the expansion process should proceed as normal.
We then applied this method to mouse brain tissue to further extend its scope. Although tissue samples pose challenges like reduced permeability, increased thickness, and heterogeneous mechanical strength, the mouse brain samples were successfully imaged using this method (Figure 2D). Similar to cell samples, protein-rich structures including cell nuclei, blood vessels, and neuronal processes were observed (Figure 2D, arrowhead). The limitation of brain tissues is that only 3.4 times expansion was achieved, which makes the effective resolution in brain samples 99 nm22. We validated the structural origin of SRS signals by correlative dye and antibody staining, in which DAPI stains for nuclei and lectin stains for blood vessels (Figure 2E,F). Neuronal cell bodies and processes were also delineated by immunofluorescence from NeuN and MAP222. With the trained convolutional neural network (CNN) algorithm utilizing the correlative fluorescence images as ground truth, the single-channel images were then segmented into specific protein-structure channels for multiplex images22.
Finally, we aimed to interrogate pathologic Aβ plaques in the brains of 5xFAD mice, a well-known animal model for Alzheimer's disease31. After following the procedures, a 3-dimensional SRS image of amyloid plaques deposition in brain tissues was acquired (Figure 2G). Puncta with high protein concentrations were observed (Figure 2G, orange arrow), representing the core of the Aβ plaque. Such image also revealed the peripheral Aβ plaques (Figure 2G, magenta arrowhead), which are often neglected by conventional Congo red staining that only targets the Aβ core. When combined with the trained segmentation algorithm, the label-free image could be transformed into a target-specific multiplex image (Figure 2F) and can be performed jointly with immunofluorescence23 to study plaque-astrocyte and plaque-microglia microenvironment interactions32 in a comprehensive and high-throughput manner.
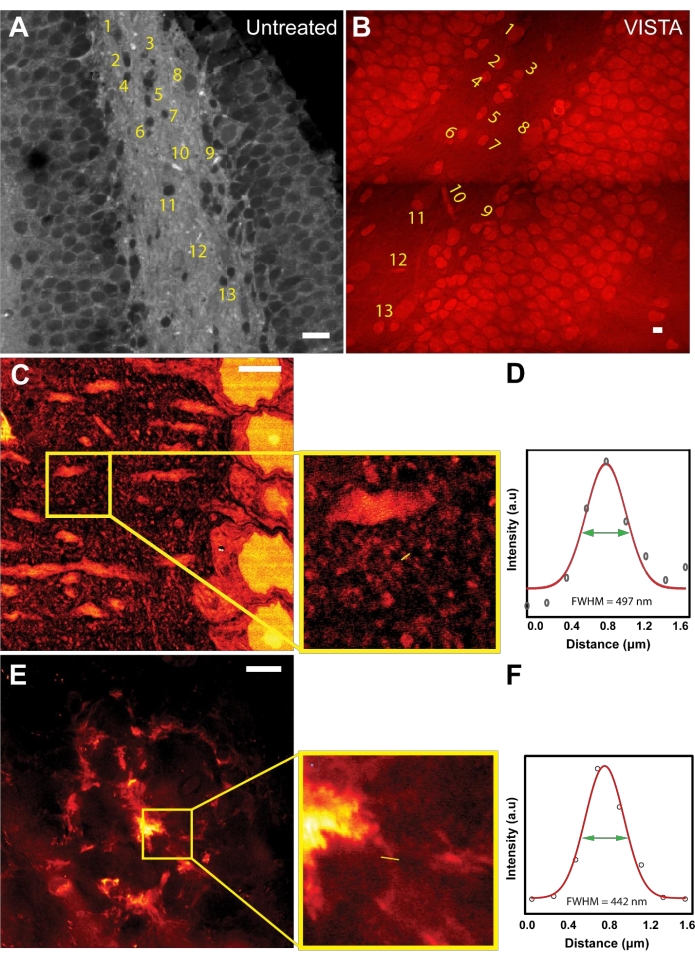
Figure 1: Sample expansion strategy enables super-resolution label-free imaging in mouse brain tissues. (A) An SRS image at CH3 frequency of mouse hippocampus. (B) A VISTA image in the same field of view of mouse hippocampus. Labeled area shows the corresponding features before and after treatments. (C) A VISTA image of a normal mouse cortex that shows finer features. Inset shows the region of interest. (D) Resolution quantification for the fine structure observed in the expanded sample. FWHM of 497 nm corresponds to an effective resolution of 146 nm with 3.4 times expansion. (E) A VISTA image of an amyloid-beta (Aβ) plaque in mouse brain tissues. Inset shows the enlarged region of interest. (F) Resolution quantification for the extrusion fiber structure of the expanded amyloid-beta plaque. FWHM of 442 nm corresponds to an effective resolution of 130 nm with 3.4 times expansion. Scale bars = 20 µm. Please click here to view a larger version of this figure.
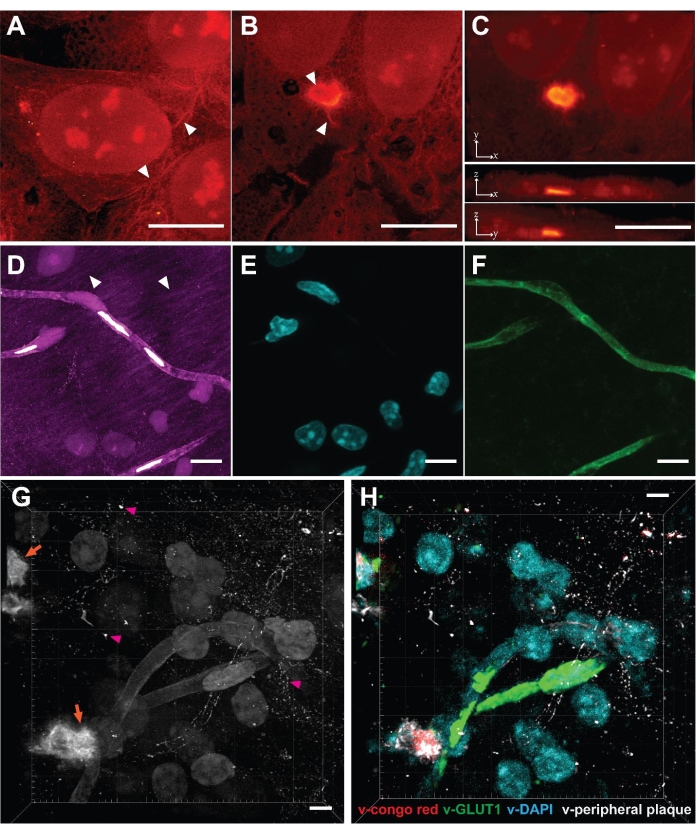
Figure 2: Label-free super-resolution volumetric imaging in cells and tissues enabled by VISTA. (A) Volumetric image of a normal HeLa cell. Arrowhead: cytoskeleton-like structure. (B) Single z-slice image of a polyQ aggregate expressed in HeLa cells. Arrowhead: hollow structure and fibril extrusions. (C) Maximum intensity projections of x-y, x-z, and y-z directions show the volumetric view of the polyQ aggregate-containing cell. (D) Volumetric image of a coronal section of mouse brain. Arrowhead: neuronal processes. (E) Fluorescence image of nuclei (DAPI staining) at the same sample region shows 1 to 1 correlation with nuclei in the VISTA image. (F) Fluorescence image of blood vessels (anti-lectin) at the same sample region shows a 1 to 1 correlation with vessel structures in the VISTA image. (G) Volumetric image of Aβ (orange arrow) containing brain tissue. Pink arrowhead: peripheral Aβ plaque. (H) Multiplex image from (G), predicted by the trained image segmentation algorithm. v-congo red represents the core of the Aβ plaque; v-GLUT1 represents blood vessels; v-DAPI represents nuclei; v-peripheral plaque represents the Aβ plaque not stained by the Congo red dye. Scale bars = 10 µm. The length scale is in terms of distance before expansion (adjusted for different expansion ratios). Please click here to view a larger version of this figure.
