All compounds in this study were characterized by 1H and 13C NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS) to confirm the product structure and assess purity. Key data for representative compounds are described in this section.
Spectral data are in good agreement with the triazole structure of 2a (Figure 3). In the 1H NMR spectrum of 2a the characteristic C5 proton of the triazole appears at 8.45 ppm as a singlet integrating for 1H. The mass spectrum obtained via ESI-MS generally shows both the MH+ peak and a M-N2 peak corresponding to the loss of dinitrogen.
Synthesis of N-(2-alkoxyvinyl)sulfonamide phthalan 3a via our protocol reliably delivers the product in >90% yield, however, substantial deviation in key parameters such as time, temperature, and heating method significantly impact the efficiency of the reaction (vide infra) and, consequently, the quality of spectral data. Figure 4a depicts the 1H NMR spectrum of pure 3a following a successful experiment. Notable is the absence of the triazole C5 proton peak around 8.5 ppm (cf. Figure 3) and appearance of two doublets at 6.07 and 6.25 corresponding to the vinyl and NH protons, respectively. In the 13C NMR spectrum of 3a, a key resonance is observed at 94.9 ppm that corresponds to the exocyclic vinyl carbon. For comparison, Figure 5 illustrates the 1H NMR spectrum of 3a decomposition products resulting from rapid degradation in CDCl3.
Figure 6 and Figure 7 show 1H/13C NMR and mass spectra that are in good agreement with the structures of reduction products 4 and 5, respectively. The 1H NMR spectrum of 4, which maintains the bicyclic phthalan substructure, shows key signals corresponding to diastereotopic methylene protons 3.49 and 3.14 ppm. Contrastingly, the 1H NMR spectrum phenethylamine 5 displays the same methylene as a simple quartet at 3.27 ppm due to free rotation in the ring-opened product resulting in first-order splitting patterns.
For compounds 6a-e, a characteristic 13C NMR signal corresponding to the ketal, hemiketal or thioketal carbon is found between 95 – 110 ppm, such as the peak at 110.0 ppm observed in the 13C NMR spectrum of 6c (Figure 8). Additionally, mass spectra obtained by ESI-MS typically show a relatively small MH+ peak along with a larger M-RX elimination fragment peak (RX = the corresponding alkoxy or thioalkyl group of 6).
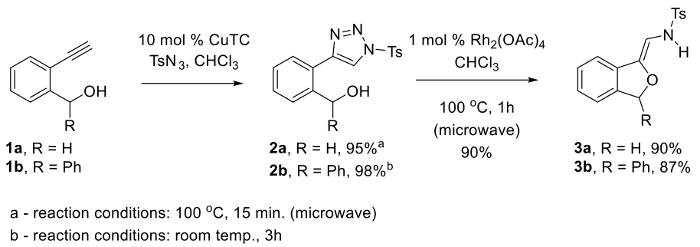
Figure 1. Synthesis of N-tosyl-1,2,3-triazoles 2 via Cu(I)-catalyzed azide-alkyne [3+2] cycloaddition and subsequent conversion to N-(2-alkoxyvinyl)sulfonamide phthalans 3 via Rh(II)-catalyzed alcohol cyclization. Please click here to view a larger version of this figure.
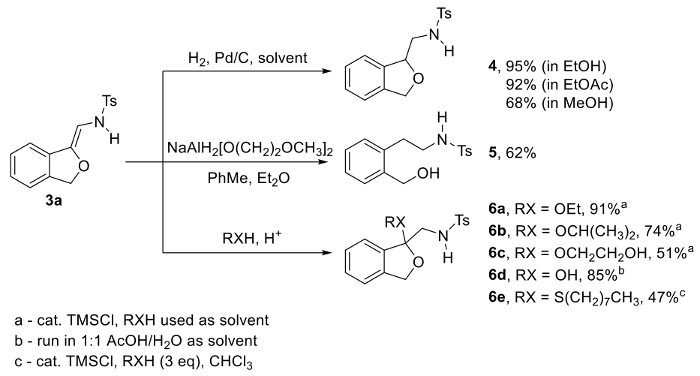
Figure 2. Differential reactivity of N-(2-alkoxyvinyl)sulfonamide phthalan 3a: conversion to reduced phthalan 4 via Pd-catalyzed hydrogenation, conversion to phenethylamine 5 via aluminum hydride reduction, and conversion to ketals 6a-c, hemiketal 6d, and thioketal 6e via acid-promoted addition of alcohols, water, and a thiol, respectively. Please click here to view a larger version of this figure.
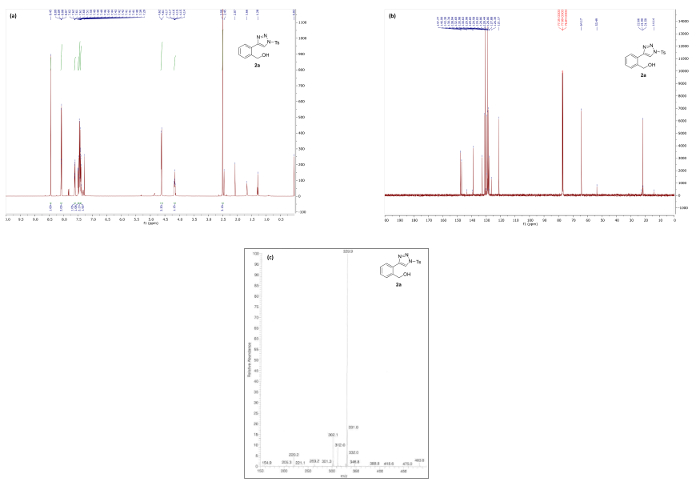
Figure 3. Spectral data for triazole 2a: (a) 1H NMR spectrum; (b) 13C NMR spectrum; and (c) mass spectrum. Please click here to view a larger version of this figure.
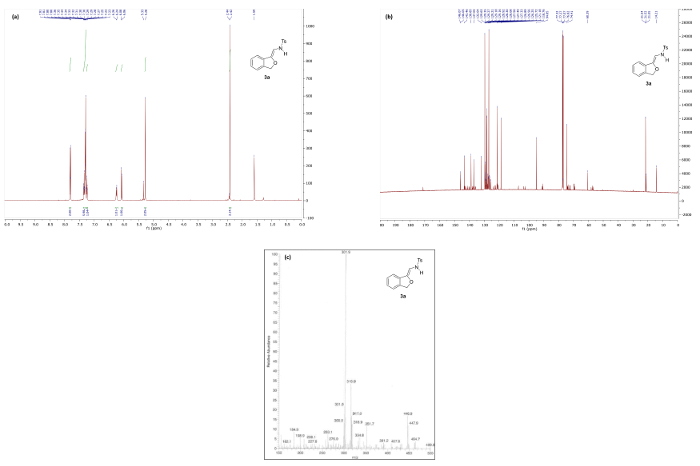
Figure 4. Spectral data for N-(2-alkoxyvinyl)sulfonamide phthalan 3a: (a) 1H NMR spectrum; (b) 13C NMR spectrum (minor peaks are decomposition products resulting from rapid degradation in CDCl3); and (c) mass spectrum. Please click here to view a larger version of this figure.
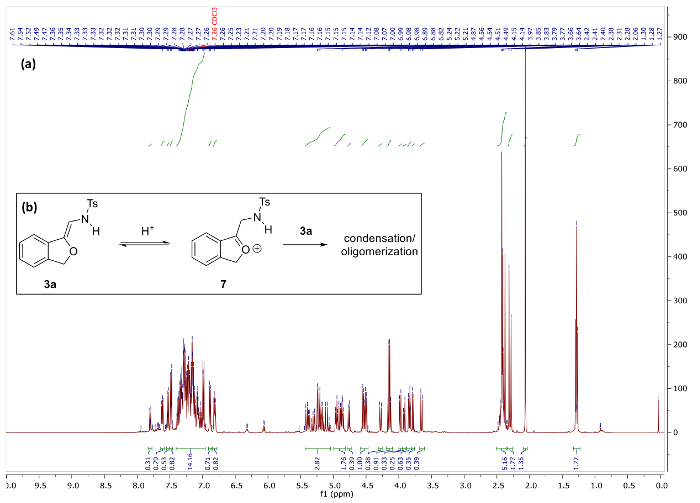
Figure 5. (a) 1H NMR spectra of 3a and decomposition products after storage in CDCl3 for 1h. (b) Hypothesized decomposition mechanism of 3a. Please click here to view a larger version of this figure.
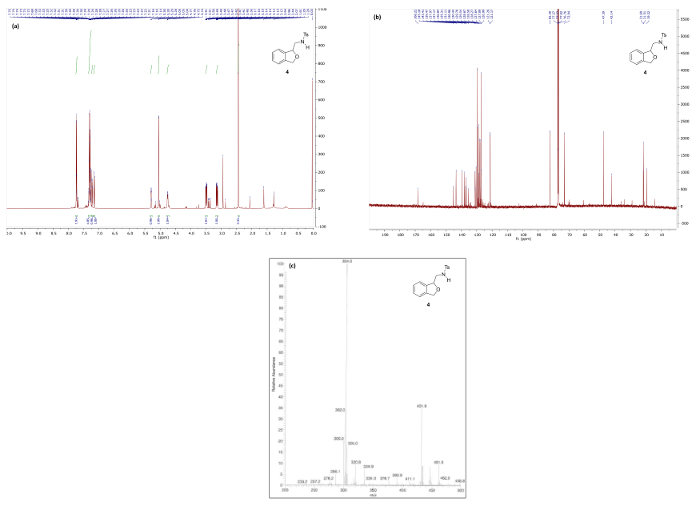
Figure 6. Spectral data for reduced phthalan 4: (a) 1H NMR spectrum; (b) 13C NMR spectrum; and (c) mass spectrum. Please click here to view a larger version of this figure.
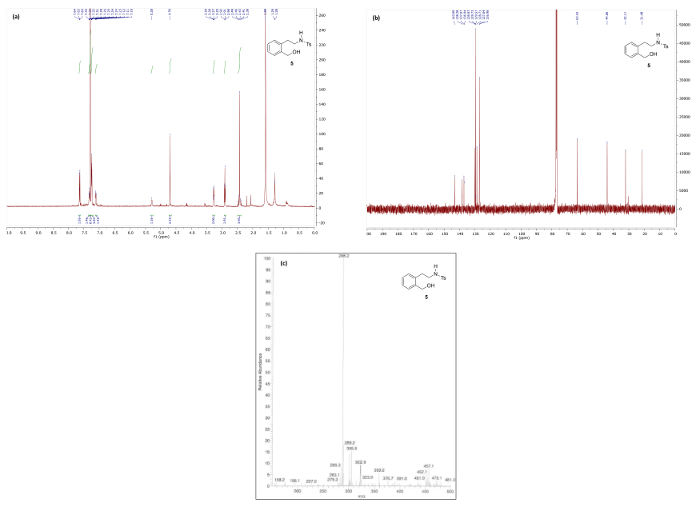
Figure 7. Spectral data for phenethylamine 5: (a) 1H NMR spectrum; (b) 13C NMR spectrum; and (c) mass spectrum. Please click here to view a larger version of this figure.
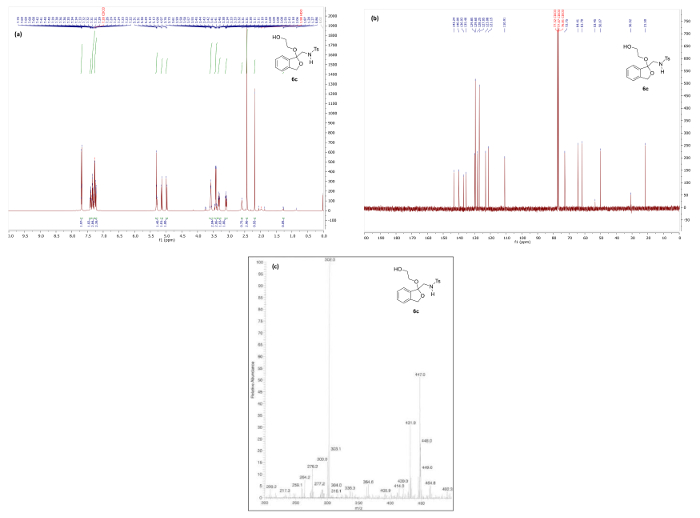
Figure 8. Spectral data for ketal 6c: (a) 1H NMR spectrum; (b) 13C NMR spectrum; and (c) mass spectrum. Please click here to view a larger version of this figure.
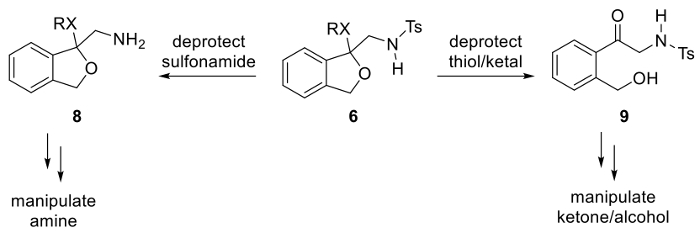
Figure 9. Strategic considerations for manipulation of compound 6, a differentially protected α-aminoketone. Please click here to view a larger version of this figure.
