Axonal transport in DA9 neuron
Using the wyIs251 line, both the anterograde and retrograde axonal transport of GFP::RAB-3 can be simultaneously recorded in the DA9 motor neuron. The average speed of anterograde and retrograde transport in the proximal dorsal axon of the DA9 neuron is about 1.8 and 2.6 μm/s, respectively22. The number of moving vesicles is about 0.03 and 0.018 per μm of axon per s. Thus, for a 30 s observation of 10 μm of DA9 axon using a 100x lens, one can find about 9 and 5 moving vesicles in the axon (Figure 4 and Movie 1). If no vesicular movement can be observed, it is possible that the laser power is too weak. In addition to long range runs, short range movement of vesicle pools can be recorded (Movie 1). Some vesicles stop at these vesicle pools, while others dissociate from there7,23. These parameters can be monitored and used to analyze mutant phenotypes.
IFT in tail neurons
Cilia are localized to the tip of dendrites in C. elegans (Figure 2B). The mnIs17 strain expresses GFP-tagged OSM-6, one of the IFT subunits29. OSM-6 is expressed in both head and tail neurons19. While many head cilia are labeled by mnIs17, only two cilia are clearly observed in the tail neuron. Thus, observing these cilia is easier than head cilia (see also Discussion). OSM-6::GFP is diffuse in the neuronal cytoplasm such as that found in the axon, cell body, and dendrite. However, in cilia, OSM-6::GFP is concentrated to cilia and incorporated in moving particles. The length of PHA and PHB cilia are about 6.5 μm. Anterograde IFT is biphasic 8,9. The speed of anterograde IFT is about 0.7 and 1.3 μm/s in the middle and distal segments of the cilia, respectively (Figure 5 and Movie 2).
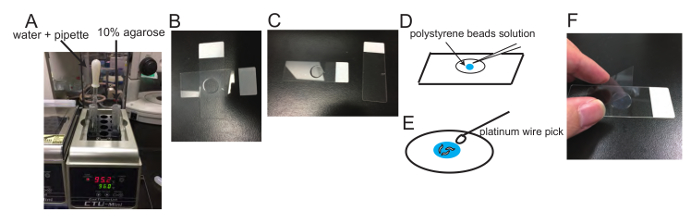
Figure 1: Demonstration of the sample preparation. (A) Hot DW, Pasteur pipet and 10% agarose on heat block. (B and C) Preparation of agarose pads: An agarose pad is formed between slides (B). The upper slide is removed after an agarose pad forms (C). (D) Schematic drawing showing how polystyrene bead solution is put on the agarose pad. (E) Schematic drawing showing how worms are put in the polystyrene bead solution using a platinum wire pick. (F) A coverslip (22 x 44 mm2) is put on the agarose pad. Please click here to view a larger version of this figure.

Figure 2: Imaging area. (A) Schematic drawing of DA9 and VA12 neurons and a representative image of wyIs251. In DA9 neurons, the ventral axon, commissure, and proximal axon does not have mature synapses. But synaptic vesicle precursors are transported from the cell body to synapses via these axonal regions. The region that should be observed is shown by boxes. (B) Schematic drawing of PHA and PHB neurons and their cilia. The region that should be observed is shown by boxes. Scale bar = 10 μm. Please click here to view a larger version of this figure.
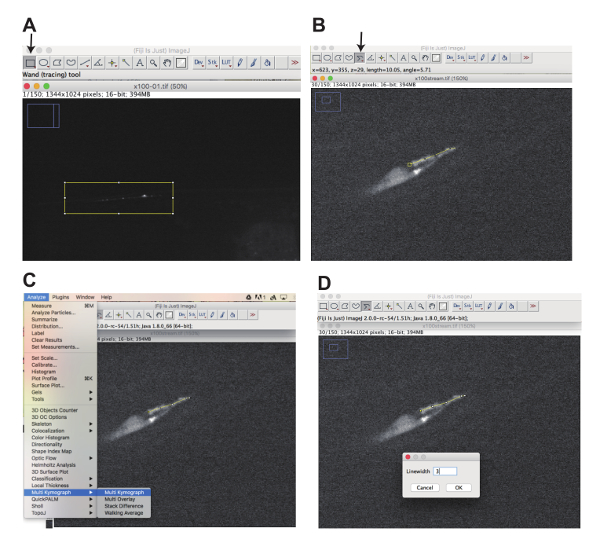
Figure 3: Generation of movies and kymographs using Fiji software. (A) Click the "Rectangular selection" button (arrow) and select the area one would like to focus on by drawing a rectangle (Yellow box) to generate substacks. (B) Click the segmented line button (arrow) and draw a segmented line along the cilia using a mouse (Yellow line). (C) Select "Analyze", "Multi Kymograph", and "Multi Kymograph" to start a plugin to generate kymographs. (S) Input linewidth, click "OK" and generate kymographs (Figure 5 and Figure 7). Please click here to view a larger version of this figure.

Figure 4: Representative kymograph of axonal transport of synaptic vesicle precursors. GFP::RAB-3 movement is observed at the proximal asynaptic region of DA9 neuron and the kymograph was generated with Multi Kymograph of Fiji. Horizontal and vertical bars represent length and time, respectively. Please click here to view a larger version of this figure.
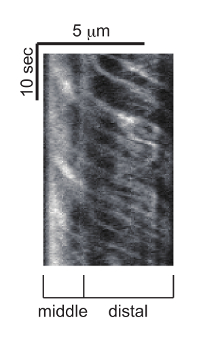
Figure 5: Representative kymograph of IFT. OSM-6::GFP is observed in PHA and PHB cilia and this kymograph is generated from the trafficking event in either one of them. The kymograph was generated with Multi Kymograph of Fiji. Horizontal and vertical bars represent length and time, respectively. Please click here to view a larger version of this figure.

Movie 1: Representative movie of axonal transport of synaptic vesicle precursors. GFP::RAB-3 movement is observed at the proximal asynaptic region of the DA9 neuron. GFP::RAB-3 is incorporated into synaptic vesicle precursors and transported. Both anterograde and retrograde axonal transport are recorded. Some vesicles stop but restart again. The movie was recorded at 4 frames/s and plays at 15 frames/s. Scale bar = 5 μm. Please click here to view this video. (Right-click to download.)

Movie 2: Representative movie of IFT. OSM-6::GFP is observed in PHA and PHB cilia. OSM-6::GFP is incorporated into the IFT complex and moves along the cilia. The movie was recorded at 4 frames/s and plays at 15 frames/s. Scale bar = 5 μm. Please click here to view this video. (Right-click to download.)
