Optimal Conditions for Injection:
Groups of zebrafish larvae were injected at 24, 48, 72, 96, and 120 hpf, at different anatomical sites, and their survival was examined every day for 5 days. After 5 days post injection, embryos injected at 24 hpf had 6.25% (2/32) survival, whereas 95% (38/40) of larvae injected at 48 hpf survived. As a control, larvae were injected with 1x PBS as a vehicle. There were no differences in survival between vehicle-injected and parasite-injected larvae, indicating no parasitic-dependent effects on survival rate of the fish (p = 0.08). Larvae injected between 72-120 hpf had comparable survival rates to 48 hpf-injected larvae at constant injection volume. For all procedures presented here, 48 hpf larvae were used due to their ease of manipulation and having developed organs and easily penetrable skin without evident damage after injection.
Larvae injected at 48 hpf were injected in the pericardial space, tail muscle, hindbrain ventricle, otic vesicle, notochord, and duct of Cuvier in the yolk sac. There were no differences in the survival of larvae injected at differing anatomical sites. However, the fastest and easiest region to inject was the duct of Cuvier located in the anterior part of yolk sac (Figure 1, Movie 1). Injections at that site allowed the introduction of higher volumes with a lower risk of injury to vital structures. Additionally, between 24-72 hpf, this region is an optimal site to directly access the developing vasculature and heart11.
Parasite Visualization Using LSFM:
Within 8-10 min following injection of T. cruzi into the duct of Cuvier, parasites were identified in zebrafish larvae using LSFM due to their CFSE fluorescent signal and the optical transparency of the larvae. After inoculation, parasites were observed either adhered to walls around the circulatory system or traveling in the direction of blood flow (Figure 2, Figure 3). When a parasite remains attached to a cardiac structure, such as the atrioventricular valve, it oscillates with heart contractions, indicating that the molecular mechanisms for the adherence of parasites might be effective in our vertebrate model ( Movie 2, Movie 3, Supplemental movie 1). T. cruzi also adhered to the walls of the larval yolk sac ( Figure 2, Movie 2), a structure which will later be reabsorbed and become part of the zebrafish intestine22. This could be similar to what happens during the chronic disease phase in infected humans, where parasites are found in cardiomyocytes and in the digestive nervous system23,24. When not attached, the parasites drifted through the blood flow in the same direction as the erythrocytes ( Figure 3, Movie 4). Parasites could be observed in different sized vessels of the fish, but were more abundant in the pericardial space and in the adjacent yolk region containing blood flow ( Figure 2, Figure 3, Supplemental movie 2).
At 10 min post injection, it was more difficult to spot single forms of the parasite due to their distribution along the vasculature, and an inability to quickly screen different anatomical sites of the fish due to a limited field of view of the LSFM (at 40X magnification). After 24 h post injection (hpi), the CFSE signal starts to accumulate in the region near the developing intestine (Supplementary figure 1).
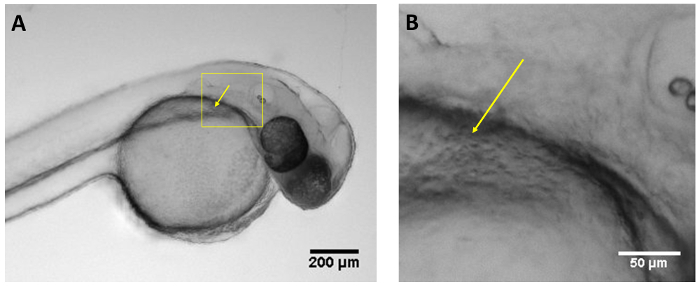
Figure 1: Optimal injection site. (A) Image of larva 48 hpf showing the optimal injection site at the duct of Cuvier (yellow arrow) using a regular stereoscope. (B) Magnified view of box in A showing the duct of Cuvier (yellow arrow). Scale bar = 200 µm (A), 50 µm (B). Please click here to view a larger version of this figure.
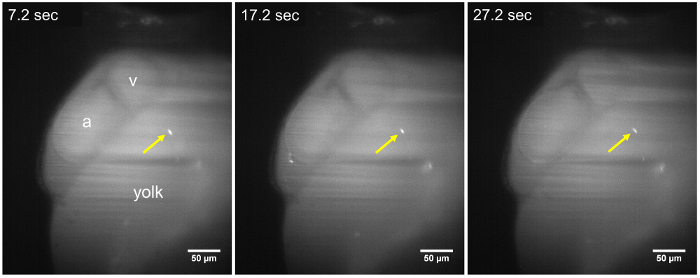
Figure 2: LSFM images of a static parasite in a 48 hpf larva. The T. cruzi parasite (yellow arrow) remains adhered to the walls of the yolk sac, throughout the time-lapse sequence (7.2 s, 17.2 s, and 27.2 s), about ~15 min after parasite injection. No change in position of the parasite is observed during an acquisition period of at least 30 s. a, Atrium; v, Ventricle. Scale bar = 50 µm. Please click here to view a larger version of this figure.
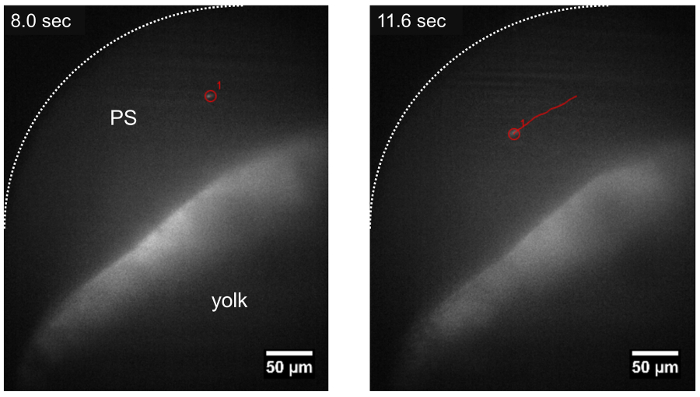
Figure 3: Trajectory of a parasite traveling in the pericardial space using LSFM. The T. cruzi parasite can be tracked while drifting in the pericardial space (PS), following the direction of blood flow (track shown in red) about ~15 min after parasite injection. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Movie 1: Blood circulation valley of the yolk in a larva 48 hpf. Movie of a larva 48 hpf showing the blood circulation valley or duct of Cuvier using a regular stereoscope. Different regions are focused during the video to show red blood cells circulating throughout the duct. Yellow arrow shows the optimal injection site. Movie was recorded about 10-15 min after parasite injection. Please click here to download the video.
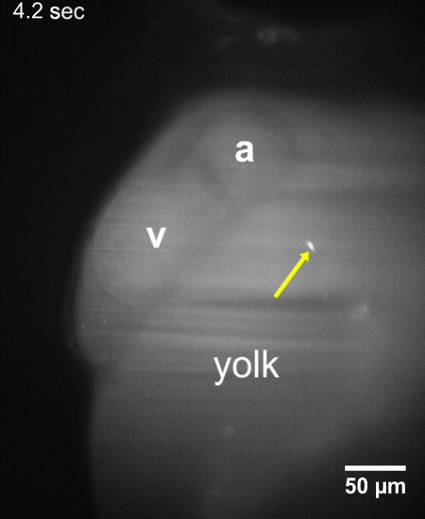
Movie 2: T. cruzi parasites attached to walls of the yolk sac. LSFM movie of a larva 48 hpf showing that T. cruzi parasites remain adhered to the yolk sac about 10-15 min after parasite injection. No change in the position of the parasite was observed during an acquisition period of at least 30 s. a, Atrium; v, Ventricle. Please click here to download the video.

Movie 3: T. cruzi parasites attached to the walls of the heart. LSFM movie of a larva 48 hpf showing that T. cruzi parasites remain adhered to the cardiovascular wall, despite the strong heart contractions about 10-15 min after parasite injection. Erythrocytes can be observed as black rounded shadows. Please click here to download the video.

Movie 4: Parasites moving in the pericardial space. LSFM movie of a larva 48 hpf showing T. cruzi parasites drifting in the pericardial space. Two parasites can be traced at different time points (ID 1, in red circle, and ID 2 in yellow circle), following a similar trajectory. The movie was recorded about 10-15 min after parasite injection. Please click here to download the video.
Supplemental Figure 1: Accumulation of CFSE fluorescent signal in the yolk. Stereoscope images of a wildtype larva injected at 48 hpf at the duct of Cuvier. CFSE fluorescent signal progressively accumulates in the yolk, after two days post injection (48 hpi). Scale bar = 500 µm. Please click here to download the figure.
Supplemental Movie 1: Parasites attached to walls and valves of the circulatory system. Stereoscope time series images of a wild type larva injected at 48 hpf. Images are taken at 0.2 s intervals capturing T. cruzi parasites moving in synchrony with cardiac muscle contractions at the atrioventricular valve. Movie was recorded about 30 min after parasite injection. Please click here to download the video.
Supplemental Movie 2: Moving and adhered parasites in the periventricular space and yolk. LSFM movie of a larva 48 hpf showing T. cruzi parasites drifting or adhered to the pericardial space or the yolk. A transmitted light view was observed for the first 5 s. The fluorescence view was observed from 5.2-25.8 s. The movie was recorded about 10-15 min after parasite injection. Please click here to download the video.
