Candida species are commensal fungi that colonize the intestinal and genitourinary tracts of all humans. Under conditions of immunodeficiency, such as that occur with premature birth or immunosuppressive effects from treatments for cancer, Candida species can become opportunistic pathogens. Of the Candida species, Candida albicans is the most prevalent fungal colonizer and causes the majority of invasive fungal infections. Other Candida species such as C. glabrata, C. parapsilosis, C. tropicalis, and C. kruseii also cause serious infections in immunocompromised patients, with some exhibiting intrinsic resistance to commonly used anti-fungal antibiotics such as fluconazole and amphotericin B. Hence, infections with some of these species are being observed more frequently, especially in patients being treated prophylactically with anti-fungal agents. Even with appropriate and timely anti-fungal treatment, invasive Candida infections continue to be associated with significant morbidity and mortality1. Because of the significance of Candida species in human health, there is a need for readily available molecular tools that allow the study and elucidation of their pathogenesis mechanisms.
One important tool that allows researchers to visualize and quantify microbial cells and the proteins that they express is FP fusion technology. Polymerase chain reaction (PCR)-mediated gene modification, as described in this paper, allows the construction of fusions, between FP sequences and a Candida protein coding sequence of interest at its genomic locus. Stable integration of the construct facilitates analysis of protein expression as well as protein localization dynamics. Plasmids containing FP sequences, optimized for expression in Candida albicans and that can be used in the PCR-mediated gene modification strategy, have been previously constructed2,3,4,5. Plasmids contain FP transformation "cassettes": a FP sequence linked to a nutritional marker gene that facilitates the transformation of C. albicans and C. parapsilosis2,3,4,5,6,7. Currently available plasmids contain a variety of selectable nutritional marker genes (URA3, HIS1, ARG4) for transformation of auxotrophic strains as well as a dominant drug resistance marker (NAT1), which facilitates transformation of clinical strains lacking auxotrophies. In addition, plasmids contain options for up to four different FP sequences (green [GFP], yellow [YFP], cyan [CFP], and cherry [mCherry]) and either an ADH1 termination sequence for construction of carboxy-terminus protein fusions, or a promoter sequence for construction of amino-terminus protein fusions. Primers are designed with homology to the plasmid DNA surrounding the FP cassette. In addition, the primers also contain 5'-extension sequences bearing homology to the yeast gene of interest to be tagged, which facilitates integration of the cassette into the genomic locus via homologous recombination (Figure 1). Gene-specific FP cassettes are generated by PCR and then transformed into Candida cells made competent for uptake of DNA by treatment with lithium acetate.
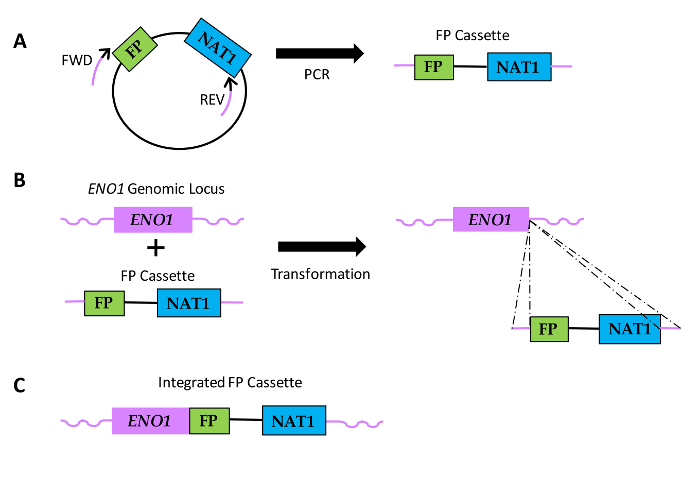
Figure 1: Diagram of how FP sequence fusions are generated in Candida species. (A) Plasmid DNA includes a FP sequence and a sequence encoding nourseothricin resistance (NAT1). Relative locations of Forward (FWD) and reverse (REV) primers are shown, with black portions of the primers indicating the region of homology to the plasmid sequence and the purple portions denoting the gene-specific homology region or primer extension. (B) FP cassettes are transformed into Candida and integrate within the ENO1 genomic locus via homologous recombination (dotted lines). (C) Resulting FP fusion sequence at the 3'end of ENO1. Please click here to view a larger version of this figure.
Herein, we present an example of protein fusion (Eno1-FP) constructions in Candida species. We use tagging plasmids containing the NAT1 transformation marker gene along with sequences encoding GFP, YFP, or mCherry (Figure 2). These plasmids are used along with primers in PCR to generate gene-specific cassettes that facilitate fusion of FPs to the 3'-end of ENO1, resulting in expression of Eno1 fused to FPs at its carboxy-terminus.

Figure 2: Maps of FP cassette-containing plasmids. Forward (F) and reverse (R) primers used to generate the cassettes from the plasmids are indicated along with the relative location of their homology to the plasmids. Primer sequences are as listed in Table 1. F1 and R1 were also used to generate the pYFP-NAT1 cassette. The plasmid containing the YFP-NAT1 cassette (pMG2263) is identical to pMG2120 with the exception of YFP in place of the GFP sequence. Cassette sizes: GFP-NAT1, 3.7 kbp; mCherry-NAT1, 3.2 kbp; YFP-NAT1, 3.7 kbp. This figure has been modified from Gerami-Nejad, et al.4 Please click here to view a larger version of this figure.
As an example, we used the protocol described above to construct GFP and mCherry fusions to Eno1 in a C. parapsilosis laboratory strain. Each putative transformant was initially restreaked for growth. In this example, since the resultant fusion protein is highly expressed (enolase) and the FPs are bright, we were able to screen transformants by fluorescence microscopy prior to performing diagnostic PCR (Figure 3)6.
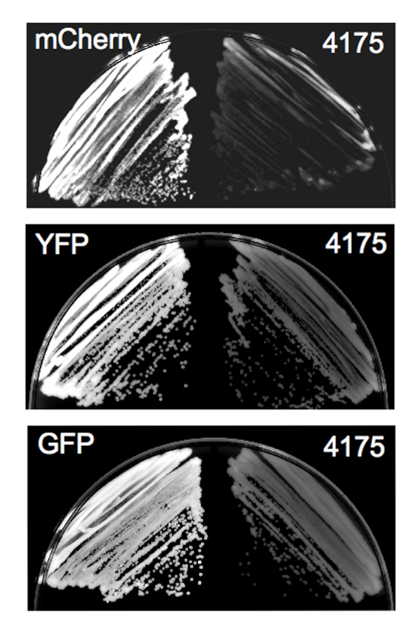
Figure 3: Colony fluorescence exhibited by Candida transformants. Representative images of yeast plate growth obtained using a laser scanning system equipped with a Cy3 filter to image the mCherry-expressing strain and a Cy2 filter to image the YFP- and GFP-expressing strains. Left panel in each pair, FP-expressing strains; right panel in each pair, non-transformed parent strain (4175) imaged with the same filter as the corresponding FP-expressing strain. Following visualization, the images were inverted and brightness and contrast were adjusted identically for FP-expressing and control strains. This figure is reprinted with permission from Gonia, et al.6 Please click here to view a larger version of this figure.
Genomic DNA was isolated9 from each of these putative transformants and used as the DNA template, along with primers complementary to sequences flanking the sites of cassette integration (i.e. sequences outside of where F and R primers anneal within the intended genomic locus to be tagged), in diagnostic PCRs to verify correct integration of the FP cassette just prior to the stop codon of the gene of interest. Of note, for lithium acetate transformations with C. parapsilosis, we observed lower efficiency (~1-2% of colonies screened contained a correct integration) as compared to our experience with C. albicans transformations.
To analyze enolase localization, we observed yeast cells expressing Eno1-FP constructs by fluorescence microscopy using a photomicroscope equipped with a 100 W mercury lamp and epifluorescence illumination with GFP, YFP and Texas Red filter sets. A charge-coupled device (CCD) camera was used to collect images that were processed with image analysis software (Figure 4A)6. In addition, we found that different FP colors were useful to visually distinguish different yeast strains within a mixture (Figure 4B)6. Because enolase is highly expressed and located in a large portion of the yeast cell, FP fusions to it can be used to generate fluorescent yeast strains that can be easily visualized, for example, in analyses of Candida-host interactions.
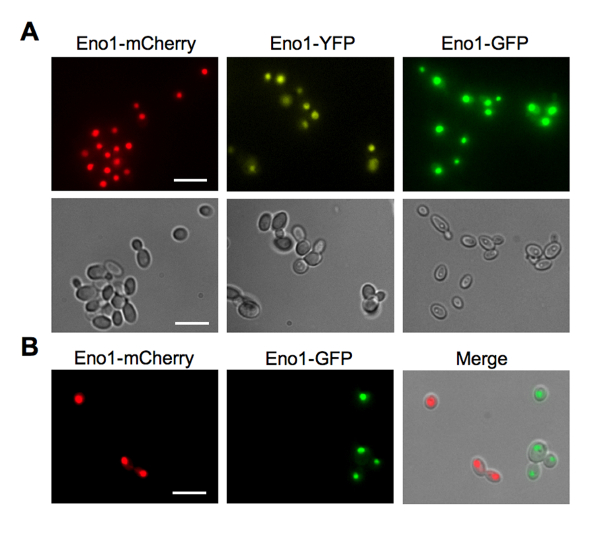
Figure 4: Expression and localization of Eno1-FP fusions by fluorescence microscopy. (A) Fluorescence (top) and differential interference contrast (DIC, bottom) images for Candida strains expressing Eno1-FPs. Eno1-FPs in C. albicans and C. parapsilosis tend to concentrate in the nucleus as well as in the cytoplasm, but to a much lower degree. (B) Microscopic images of a mixed culture of Candida strains expressing Eno1-mCherry and Eno1-GFP. Right panel, Texas red filter; center panel, GFP filter; right panel, merge of both fluorescent panels with the DIC image. Scale bars = 10 µm. This figure is reprinted with permission from Gonia, et al.6 Please click here to view a larger version of this figure.
FP fusions also facilitate analysis of protein expression levels by Western blotting. Using yeast cells generated by the protocol described above, proteins were isolated from cell lysates, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene difluoride (PVDF) membrane10. Eno1-FP fusions were detected on the blots using commercially available antibodies as previously described6. Briefly, mouse anti-GFP (1:2,000 dilution), followed by horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000 dilution) antibodies were used to probe for GFP- and YFP-tagged enolase. Rabbit anti-mCherry (1:2,000 dilution) followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (1:10,000 dilution) antibodies were used to probe for mCherry-tagged enolase. All fusion proteins were readily detected from lysates of successful transformants (Figure 5, lanes 2, 5, and 7)6 and were of expected size (~ 75 kDa) as compared to a protein ladder standard and C. albicans enolase-FP standards (Figure 5, lanes 1 and 6)6. No signal was observed for the untagged parent yeast strain (Figure 5, lanes 3 and 4)6.
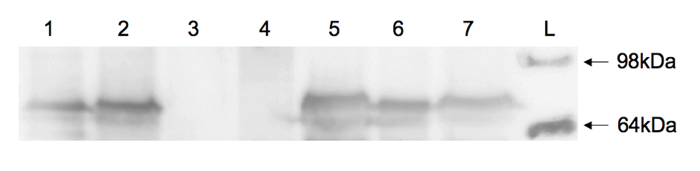
Figure 5: Immunoblot of cell lysates from Candida strains. Lane 1, C. albicans containing ENO1-mCherry fusion (J. Berman, University of Minnesota) (positive control); lane 2, C. parapsilosis containing ENO1-mCherry fusion; lanes 3 and 4, untransformed C. parapsilosis parent strain 417511 (negative control); lane 5, C. parapsilosis containing ENO1-YFP fusion; lane 6, C. albicans containing ENO1-GFP fusion (J. Berman, University of Minnesota) (positive control); lane 7, C. parapsilosis containing ENO1-GFP fusion; lane L, protein ladder with 98 and 64 kDa protein standards indicated. Lanes 1-3 were incubated with anti-mCherry antibody and lanes 4-7 were incubated with anti-GFP antibody. This figure is reprinted with permission from Gonia, et al.6 Please click here to view a larger version of this figure.
| Protocol Step/Item | Modification | Potential Improvement (cautions) |
| Primer design | Lengthen primers by including longer region of homology to genomic locus of interest | Improve binding efficiency and homologous recombination (modifying primer length may change optimal annealing temperatures) |
| PCR (amplicon quality) | Use higher fidelity Taq Gel purify PCR product |
Reduce introduction of errors into cassette sequences to improve targeting of primer to genome sequence and transformation efficiency (gel purification may decrease amplicon quantity and hence, transformation efficiency) |
| PCR (amplicon quantity) | Increase number of PCR reactions and pool products | Increase number of transformed cells (too much amplicon can also result in reduced transformation efficiency) |
| Cassette precipitation | Perform centrifugation at 4 °C, keep all reagents and tubes on ice | Increase DNA yield and, hence, transformation efficiency |
| Heat Shock | Shorten the time | Improve viability of stressed yeast cells (shorter heat shock times could also result in decreased uptake of DNA cassette by cells) |
| Recovery Growth1 | Allow for longer recovery on YPAD agar before plating to agar containing nourseothricin | Stressed yeast cells may need longer recovery times when exposed to nourseothricin (longer incubation times may also increase contaminant and false-positive transformant growth) |
| Colony Outgrowth1,2 | Incubate plates for longer times (~ 5 d) | Stressed yeast cells may need longer incubation times for colony growth (longer incubation times may also increase contaminant and false-positive transformant growth) |
| 1 Specific to transformations using NAT1 selection marker or 2 transformations of yeast strains containing genetic mutations that affect growth | ||
Table 2: Guide for troubleshooting and optimization.
