1. Transformation of E. coli with the pGEX-RhoA(G17A) Construct
- Prepare LB-Agar by dissolving 2.5 g LB and 1.5 g Agar in 100 ml dH2O. Autoclave and cool to an estimated 50-55 °C, which as a rule of thumb, is when the flask can be held comfortably.
- Prepare Ampicillin (Amp) stock by dissolving 50 mg/ ml in dH2O. Syringe filter and freeze unused aliquots. Add 100 μl of Amp stock (final concentration 50 μg/ml) to the LB-Agar from 1.1. Swirl to mix and pour into 10 cm bacterial dishes (15-20 ml/dish). Allow it to solidify (15-30 min.) and store unused plates inverted at 4 °C for 2-3 weeks.
- To transform E. Coli, quickly thaw an aliquot of DH5α competent cells in an ice bath. Add 1 μl of pGEXRhoA(G17A) DNA diluted to 25-50 ng/μl. Flick the tube to mix and incubate on ice for 30 minutes. Heat shock at 42 °C for 45 seconds and place back on ice for 2 minutes. Add 900 μl SOC medium and grow for one hour at 37 °C with shaking.
- Spread 50-100 μl of the transformed bacteria on an LB-Agar-Amp plate using a bent sterile Pasteur pipette. Incubate the plate right side up in a 37 °C incubator for 5 minutes and then invert and grow overnight.
- A single colony will be picked from the plate for preparation of the GST-tagged protein (step 2.1). For future use, wrap and store plates inverted at 4 °C for about 3 weeks. In addition, bacterial stocks can be prepared for more prolonged storage by growing individual colonies in 2 ml sterile LB-Amp overnight at 37 °C with shaking. Mix an aliquot with sterile 80% glycerol in a 1:1 ratio and freeze at -80 °C.
2. Preparation of GST-RhoA(G17A) Beads
- Prepare LB by adding 25 g LB to 1 L dH20 and autoclaving. When cool, add 50 μl Amp from stock to 50 ml LB (50 μg/ml final concentration). Inoculate with a well isolated colony of transformed bacteria and grow overnight at 37 °C with agitation. When at full density (OD600 > 1.0) dilute with 450 ml LB-Amp and grow for an additional 30 minutes at 37 °C.
- Prepare a 100 mM stock solution of Isopropyl B-D-thiogalactopyranoside (IPTG) by dissolving 0.238 g in 10 ml dH2O. Store in aliquots at -20 °C. Induce bacteria to produce Rho protein by adding 500 μl 100 mM IPTG to 500 ml culture (a final concentration of 100 μM). Reduce temperature to 22-24 °C and grow for ~16 h hours.
- Spin culture at 3600 g for 10 minutes at 4 °C. If needed, the 500 ml culture can be divided into 50 ml tubes for centrifugation. Freeze pellet(s) for at least 1 hour (or preferably overnight) at -80 °C.
- Prepare 200 ml lysis buffer containing 20 mM HEPES (0.95 g)/ pH 7.5; 150 mM NaCl (1.75 g); 5 mM MgCl2 (0.203 g); 1% TX-100 (2 ml). Prepare stock solutions of 1M DTT (1.542 g in 10 ml dH2O) and 100 mM PMSF (0.174 g/10 ml EtOH). To prepare lysis buffer +, supplement 10 ml with 1mM DTT (10 μl of stock) and 1 mM PMSF (100 μl of stock) and one Complete Mini Protease Inhibitor tablet.
- Working on ice, add 10 ml lysis buffer+ to the pellets from step 2.3. Resuspend thoroughly by gentle vortexing and pipetting. Avoid foaming. Sonicate on ice for 1 minute at setting 4 with 50% pulse. Spin the sonicated lysate at 15,000-20,000 g for 15 minutes at 4 °C, and remove the clarified sonicate (supernatant) to a sterile capped 15 ml tube.
- Prepare the Glutathione Sepharose by gently mixing the original tube containing a 75% slurry and transfer 335 μl into a 15 ml tube. Use a wide bore tip to pipette beads. Add 10 ml cold PBS, and spin 500 g for 5 minutes at 4 °C. Discard the supernatant, add 1 ml lysis buffer+ to the beads and spin as for previous wash. Discard the supernatant and add lysis buffer+ to make a 50% slurry.
- Add 250 μl of equilibrated bead slurry to the supernatant from step 2.5. Rotate at 4 °C for 45 minutes.
- Prepare 500 ml HBS containing 20 mM HEPES (2.38 g)/pH 7.5 and 150 mM NaCl (4.38 g) in dH2O. Prepare stock solutions of 1M MgCl2 (0.952 g in 10 ml dH2O) and 1M DTT (1.542 g in 10 ml dH2O). To prepare HBS+, supplement 100 ml just before use with 5 mM MgCl2 (50 μl from stock) and 1 mM DTT (100 μl from stock).
- Spin the beads from step 2.7 at 500 g for 5 minutes at 4 °C. Discard the supernatant and wash beads 2x with 10 ml lysis buffer+, and 2x with 10 ml HBS+. After the final wash, make a 50% slurry by resuspending the beads in HBS+ supplemented with BD BaculoGold protease inhibitor (20 μl of 50x BD BaculoGold/ml).
- Dilute 10 μl of the final beads preparation with 2x Laemmli sample buffer containing β-mercaptoethanol. Make Bovine Serum Albumin (BSA) standards. Use a 2 mg/ml stock (0.02 g of BSA in 10 ml dH2O). Mix 10 μl of stock with 10 μl Laemmli (20 μg final); 5 μl of stock with 5 μl of dH2O and 10 μl Laemmli (10 μg final); and 2.5 μl stock with 7.5 μl dH2O and 10 μl Laemmli (5 μg final). Boil all samples (5 min). Spin the bead sample and run supernatant with BSA standards and molecular weight markers on a 10% SDS-polyacrylamide gel.
- Prepare the Comassie Blue stain (0.1 g in 10 ml Acetic Acid, 40 ml Methanol and 50 ml dH2O) and the destain solution (500 ml dH2O, 400 ml methanol and 100 ml acetic acid). Store at room temperature. Stain the gel for 20-30 minutes, remove the dye (it can be reused multiple times) and rinse with destain solution twice. Continue to destain with gentle shaking for several hours until bands are clearly visible.
- Estimate the concentration of GST-RhoA(G17A) coupled to the beads using the BSA standards as a reference (Fig 2). Aliquot an equal volume of beads containing ~10-15 μg protein into 1.5 ml micro centrifuge tubes. Store beads at 4 °C to use within a day. Freeze at -80 °C in HBS+/glycerol in a 3:1 ratio to use within a few days.
3. GEF Pulldown Assay with Nucleotide-free RhoA(G17A) Beads
- Grow cells in 10 cm dishes to confluence. Serum deprive for at least 3 hours and treat as required.
- Prepare lysis buffer+ as in step 2.4. Prepare enough lysis buffer for 700 μl/dish plus some extra amount to allow for pipetting errors. Add the protease inhibitors just before use.
- Working on ice, remove culture medium from the dishes and wash with ice-cold HBS. Remove all the HBS and add 700 μl lysis buffer+ to each dish. Swirl plates to cover all areas, scrape and collect lysates into numbered 1.5 ml tubes. Spin at 15,000 g for 1 min at 4 °C. The supernatant will be used for the assay.
- If your cell number is equivalent in all dishes being tested, you can omit doing a protein assay, and move to step 3.5. Otherwise measure the protein concentration of each supernatant using Bio-Rad quick protein assay and equalize the supernatant for volume and concentration. The amount of total protein depends on the cell types used (typically 1-1.5 mg protein for LLC-PK1 cells).
- Remove 30 μl of each supernatant and mix with 30 μl 2x reducing Laemmli sample buffer, boil and set aside for step 3.7. Add remaining supernatants to aliquots of the GST-RhoA(G17A) beads from step 2.12. Rotate for 45 minutes at 4 °C.
- Spin beads at 6800 g for 10 seconds at 4 °C. Discard the supernatant and wash the beads 3x with lysis buffer, spinning in the same way between washes. Completely remove the final wash using a 1 cc syringe fitted with a 30 G needle and add 20 μl 2x reducing Laemmli sample buffer. Boil for 5 min. Spin to pellet beads and either run the supernatant immediately (preferable) or store it at -80 °C for later analysis.
- Run 20 μl total cell lysates and all of the precipitated protein samples on the appropriate percentage SDS-polyacrylamide gel for the size of GEF you are studying. Detect your GEF of choice by Western blotting using a specific antibody.
4. Representative Results
Part 1 and 2 of the protocol describes preparation of GST-RhoA(G17A) coupled to GSH-sepharose beads and its testing by SDS-PAGE (see outline of protocol on Fig 1). A typical Coomassie stained gel is shown on Fig 2. The sample with the eluted protein should contain a single band at approximately 50 kDa (Fig 2, lane 6). The concentration of the protein can be estimated using the BSA reference samples. In the example on Fig 2, the concentration of RhoA(G17A) is estimated to be 15 μg/10 μl. Thus, aliquots of 10 μl/tube were prepared. The typical yield in our hand is 15-20 μg protein from 10 ml bacterial lysis. Part 3 of the protocol describes the affinity precipitation assay (see overview on Fig 1). A successful GEF assay detecting activation of the exchange factor GEF-H1 is shown on Fig 3. The RhoA(G17A) protein captured some GEF-H1 from the control (untreated) cell lysates, suggesting that GEF-H1 has basal activity. The amount precipitated however increases in cells treated with the inflammatory cytokine Tumor Necrosis Factor -α (TNF- α), consistent with the notion that TNF-α activates GEF-H1 5,7. Importantly, the total cell lysates show similar amounts of GEF-H1 in the control and the treated sample, suggesting that the treatment did not alter GEF-H1 levels and the input used in the assay is equal.
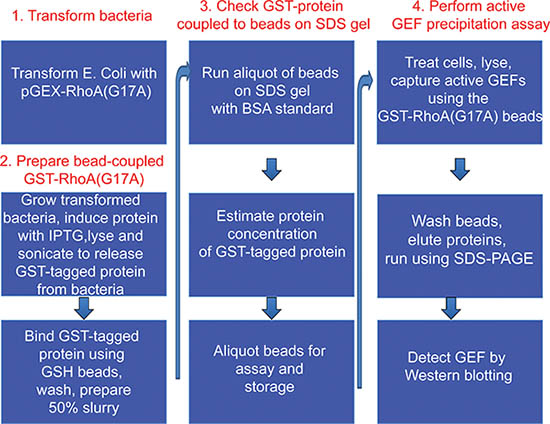
Figure 1. Overview of the protocol.
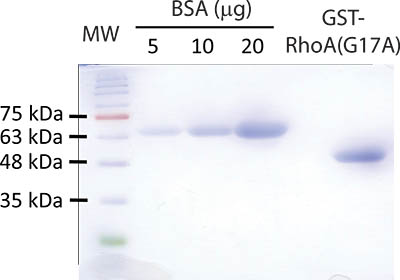
Figure 2. Representative result of the bead preparation protocol. A Coomassie stained gel with successful GST-RhoA(G17A) bead preparation is shown. Bead sample and BSA protein standards were separated by SDS-PAGE using a 10% acrylamide gel. To test the beads, 10 μl of the final bead slurry containing GST-RhoA(G17A) is diluted 1:1 with reducing Laemmli sample buffer and boiled for 5 minutes. The beads were spun briefly and the supernatant loaded on the gel. The following samples were loaded: Lane 1: molecular weight marker (MW) (FroggaBio BLUeye prestained protein ladder); Lanes 2-4: 5, 10 and 20 μg Bovine Serum Albumin (BSA); Lane 5 is empty; Lane 6: 10 μl of the freshly prepared RhoA(G17A) beads. After separation is completed, the gel is stained using Coomassie Blue and subsequently destained to reveal proteins. The molecular weight of the GST-RhoA(G17A) protein is roughly 50 kDa and runs around the level of the 48 kDa marker. The concentration of the GST-Rho protein in this particular sample is estimated to be around 15 μg/10 μl slurry.
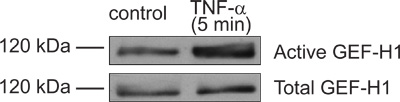
Figure 3. Representative GEF activation assay showing TNF-α-induced activation of GEF-H1. Confluent LLC-PK1 cells were treated with 10 ng/ml TNF-α for 5 minutes. Following treatment the cells were lysed and active GEFs were captured using GST-RhoA(G17A) bound beads. The presence of GEF-H1 in the precipitated proteins (top blot) and total cell lysate samples (bottom blot) was detected using Western blotting with an anti-GEF-H1 antibody (Cell Signaling). Please note the increased amount of precipitated GEF-H1 in TNF-α-treated versus non-treated cells relative to the equivalent inputs, indicating a successful result.
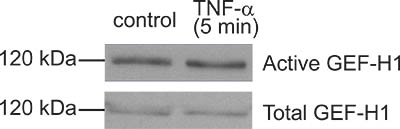
Figure 4. A “bad result”. Although the GEF pulldown assay shown here resulted in some GEF-H1 captured by the beads, the amounts precipitated from control and TNF-α-treated cells are the same. Thus TNF-α, a know activator of GEF-H1 in this case did not induce activation. In this particular experiment subsequent troubleshooting suggested that the TNF-α used was not fresh enough and probably degraded.
