Results can be visualized using appropriate software. For each well, the 1D amplitude plot displays all droplets relative to their amplitude. A clear separation between positive and negative droplets is expected. If more than 10% of the total droplets are found between the positive and negative droplet clouds (a phenomenon referred to as droplet rain), it is necessary to re-measure the sample (see Figure 1). Additional information on droplet rain is available in the discussion.
A data table can be created to summarize all recorded information, including the sample name, number of accepted droplets (event counts), and concentration (copies/µL). Ideally, the event count should fall between 15,000 and 20,000 accepted droplets. If the event count for a well is below 10,000, the data point should be excluded from the analysis. An example of the output data is provided in Table 5.
The vg titer can be calculated based on the concentration (copies/µL), with consideration of sample dilutions. The working range of dd_PCR is 10-10,000 copies/µL. Values below 10 copies/µL should be excluded from the analysis. The vg titer for samples, as well as the positive and negative controls, should be calculated. The measured value of the positive control should have a coefficient of variation (%CV) lower than 20% relative to the theoretical value. The negative control should be less than 5 copies/µL. Additionally, the %CV between repeats and different dilutions for each sample should be calculated. If the %CV between different dilutions exceeds 20%, the value may be considered inaccurate, and the sample may need to be re-measured.
A successful measurement is characterized by a clear separation of the positive and negative droplet clouds, at least 10,000 accepted droplets, a value between 10 copies/µL and 10,000 copies/µL, and a %CV between repeats lower than 20%.
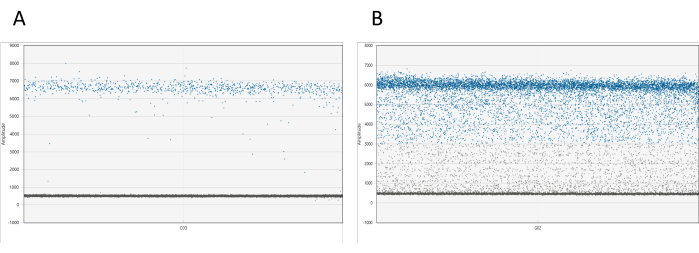
Figure 1: Visualization of dd_PCR droplets. (A) The 1D amplitude plot demonstrates a clear separation of positive and negative droplets, indicating successful droplet partitioning. (B) The plot shows a poor separation of positive and negative droplets, known as droplet rain, which suggests suboptimal partitioning or potential issues with the assay. Please click here to view a larger version of this figure.
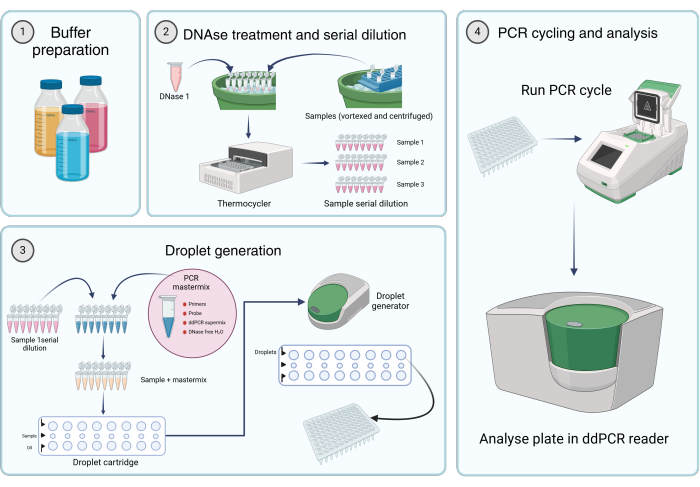
Figure 2: Workflow for quantifying viral genomes using digital droplet PCR. (1) Buffers, reagents, and solutions are prepared according to the manufacturer's instructions or step 1 of the protocol (Preparation of stock solutions). (2) 45 µL of the DNase I solution is aliquoted into each tube of an 8-tube PCR strip. After vortexing and briefly centrifuging the samples, 5 µL of the sample is added to one of the tubes. The DNase I and sample-containing eight-tube PCR strip are incubated in a thermocycler for 1 h at 37 °C, followed by serial dilutions of the samples. (3) The PCR mastermix is prepared as described, and 19.8 µL is aliquoted into each tube of an 8-tube PCR strip. Sample serial dilutions from step 2 are added to the mastermix. 20 µL of the mastermix plus
sample solution is loaded into the middle row of the cartridge, and 60 µL of droplet-generation oil is transferred to the lower wells of a droplet-generating cartridge. The cartridge is then placed into the droplet generator and run according to specified conditions. After droplet generation, 42.5 µL from the top row of the cartridge is transferred to a multi-well PCR plate. (4) The PCR plate is loaded into a PCR thermocycler and run according to the provided conditions. The plate is analyzed using a dd_PCR reader. Please click here to view a larger version of this figure.
Table 1: Precision data. This table presents precision data from four sets of quality control (QC) samples, each with five concentrations (QC1-QC5). Each QC was measured five times. The coefficient of variation (%CV) between different runs, representing repeatability (A), and between different sets of samples, representing intermediate precision (B), is shown. Please click here to download this Table.
Table 2: Sample dilutions. The table shows the recommended sample dilutions based on the expected viral genome titer (vg/mL). A total of three dilutions are recommended to ensure at least two values fall within the working range of the assay. Please click here to download this Table.
Table 3: dd_PCR master mix composition. This table outlines the composition of the dd_PCR master mix, which includes a forward and reverse primer (909 nM), probe (227 nM), and dd_PCR supermix for probes (without dUTP, 1x). Please click here to download this Table.
Table 4: Thermocycling conditions. The table details the recommended PCR program, which includes: (1) a 10 min incubation at 95 °C for capsid disruption and enzyme activation, (2) 40 cycles of 30 s at 94 °C for DNA denaturation, and 1 min at 60 °C for annealing and extension, (3) a 10 min incubation at 98 °C for enzyme deactivation, and (4) holding at 4 °C. The annealing temperature may require optimization based on the primer/probe set used. Please click here to download this Table.
Table 5: Example of output data from an actual AAV run. The table provides an example of output data from an actual AAV run, where one sample was measured in two different dilutions, each in duplicate. The viral genome (vg) titer (in vg/mL) is calculated using the following formula: 10 (DNase I pre-treatment) x 10 (dilution in the master mix) x 1,000 (µL to mL) x dilution in AAV dilution buffer. STDEV represents standard deviation, and CV indicates the coefficient of variation. Please click here to download this Table.
