The deletion constructs for all four chitinolytic genes encoded in the U. maydis genome were generated by Golden Gate cloning using the hygR cassette for deletion of cts1, the natR for deletion of cts2, the G418R cassette for deletion of cts3 and the cbxR for deletion of cts4 20. A general overview of the gene replacement strategy is exemplified by the deletion of cts3 (Figure 1). Single and double mutants of cts1 with a second chitinolytic gene were generated sequentially with the same constructs in two different genetic backgrounds for analysis of the role of chitinases in virulence. Two different strain backgrounds were employed: AB33 allows induction of filamentous growth, the first step towards pathogenicity 23, SG200 is a solo-pathogenic strain that enables rapid disease scoring 10. After transformation of the cts3-deletion construct into the appropriate strain background, single colonies of the second selection plate were tested for the correct deletion by diagnostic PCRs (Table 1) and Southern blot (Figure 3). Interestingly, for cts3 the homologous recombination rate was much higher in SG200 than in AB33, but this difference was not consistently observed in deletions of the other chitinases.
The stringent strain verification ensures a single insertion of the deletion construct at the desired position. However, additional modification such as point mutations can remain undetected, which might mislead the interpretation of phenotypes. Therefore, at least two independent transformants are phenotyped.
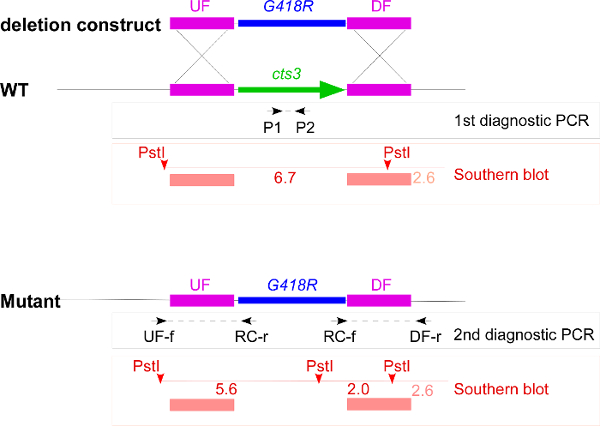
Figure 1. Overview of the deletion strategy exemplified by cts3. This schematic overview includes the deletion construct with upstream flank (UF), downstream flank (DF), and the geneticin resistance cassette (G418R), the genomic locus of wildtype (WT) and cts3 deletion mutant and all primers as well as the restriction sites used in the strain verification. The first diagnostic PCR results in a product only if the wildtype gene copy is present, the two second diagnostic PCRs result in products if the resistance cassette has been correctly integrated. For the Southern blot the genomic DNA is digested with PstI, the probes span the UF and DF (red bars) leading to detection of a 6.7 kb band in wildtype and two bands of 5.6 kb and 2.0 kb in the mutant. In addition, the DF probe weakly recognizes a 2.6 kb product. Please click here to view a larger version of this figure.
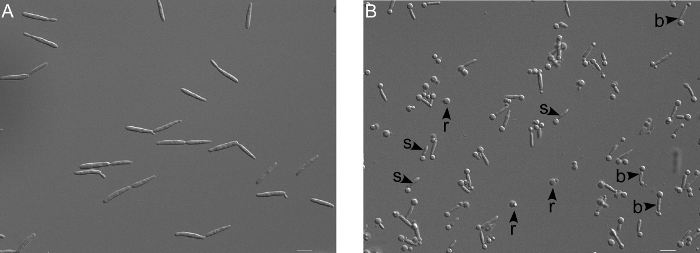
Figure 2. Microscopic verification of protoplasting reaction. (A) Untreated wildtype cells (FB2) appear cigar shaped. (B) Upon treatment with cell wall degrading enzymes (here for for 30 min), the appearance of round spheres at one end (s) and bar-bell like structures (b) indicates that the cell wall degradation has started. Finally the protoplasts are completely round (r), which is due to the lack of cell wall. Scale bar = 10 µm. Please click here to view a larger version of this figure.
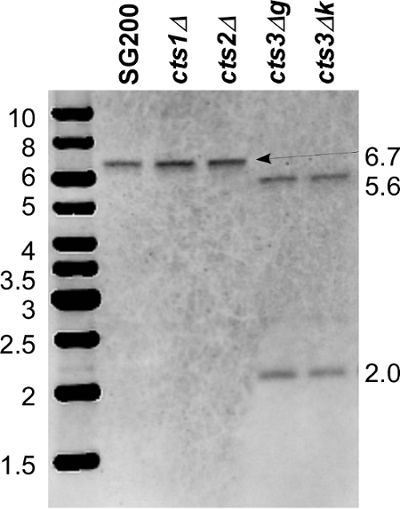
Figure 3. Southern blot for cts3 deletion. Genomic DNA was digested with PstI. Both flanks were labelled, mixed in equimolar ratios and used as a single probe. In the wildtype SG200 and deletions of the chitinases cts1 and cts2, a single band of 6.7 kb is detected. In cts3 deletions (g and k), two bands of 5.6 kb and 2.0 kb are visible indicative of the correct deletion of cts3. An additional band of 2.6 kb can be visualized by massive overexposure. This corresponds to a fragment that is recognized by an overlap of only 100 bp of the downstream flank probe. Please click here to view a larger version of this figure.
Some mutations lead to obvious growth defects that can be detected microscopically. In the chitinase deletions, single mutants did not display any obvious defect, while cts1/2 double mutants showed a pronounced cell separation defect 20. Staining of septa showed that cytokinesis was completed but the cells remained connected likely via residual chitin (Figure 4 from Langner et al. 2015). A similar microscopic growth phenotype is known for the deletion of khd4 24, a gene encoding an RNA-binding protein mediating post-transcriptional RNA turnover of genes involved in morphology and pathogenicity 25.

Figure 4. Microscopic analysis of deletion mutants. Cell morphology and septum formation of the chitinase deletion strains. cts1/2Δ strains exhibit a cell separation defect and form large aggregates, which is not due to a lack of septum formation. Primary and secondary septa (see 5x enlargement in insets) were stained with Calcofluor White (CW) prior to microscopy. Scale bars = 10 µm. This figure is reprinted with permission from Langner et al. 2015 Eukaryotic Cell 20. Please click here to view a larger version of this figure.
To test the contribution of chitinases to virulence, seedling infection assays were carried out using the mutants in the SG200 strain background. While mutants in khd4 were reduced in virulence, deletion of the chitinases did not affect infection of maize seedlings (Figure 5) 20,25.
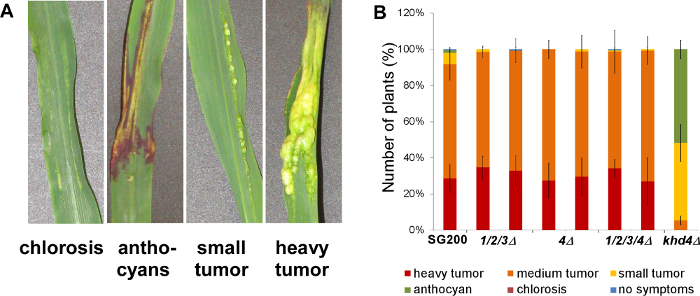
Figure 5. Infection assay of deletion mutants. Disease rating of maize seedlings 9 days after infection with the respective U. maydis strains. (A) Macroscopic symptoms that are used for disease scoring. (B) Two independent transformants were tested for each strain (N >100). All chitinase-deficient mutants (1/2/3Δ: triple mutant endochitinases, 4Δ: single mutant N-acetylglucosaminidase, 1/2/3/4Δ: quadruple mutant) infected the host plant leading to heavy tumor formation as observed in wildtype infections with the solopathogenic strain SG200. By contrast, a strain carrying a deletion of the gene encoding the RNA-binding protein Khd4 was reduced in virulence as reported previously 24. Error bars indicate standard deviation of three independent experiments. This figure is modified with permission from Langner et al. 2015 Eukaryotic Cell 20. Please click here to view a larger version of this figure.
| Genetic background | 1st diagn. PCR (excluded/tested) |
2nd diagn. PCR (confirmed/tested) |
Southern (confirmed/tested) |
% recomb. |
| AB33 | 7/22 | – | 7/15 | 32 |
| AB33 cts1Δ | 19/24 | – | 5/5 | 21 |
| SG200 | 0/10 | 8/10 | 7/8 | 70 |
| SG200 cts1Δ | 0/20 | 19/20 | 14/19 | 70 |
Table 1: Strain verification for cts3 deletions. The chitinase gene cts3 was deleted in four different genetic backgrounds to allow studies of filamentous growth (AB33) and infection (SG200) in single and multiple deletions.
