A2B5-positive OPCs are isolated through positive cell selection using a magnetic column separation system. Prior to the isolation procedure, the whole brain is removed from rat pups that are between P5 and P7. Once the whole brain is successfully detached from the skull, the olfactory bulbs and cerebellum are removed using fine surgical forceps (Figure 1A). In order to isolate cerebral cortical tissue the hypothalamus, thalamus and midbrain are excised by careful dissection (Figure 1B). Next, the hippocampus and striatum are removed using bent surgical forceps (Figure 1C–D). The isolation process efficiently eliminates meningeal and fibroblast cells, but enzymatic digestion is improved when meninges and vasculature are removed (Figure 1E). Finally, tissue blocks of 1 mm x 1 mm are prepared for the subsequent tissue dissociation steps (Figure 1F).
A good OPC isolation should result in a compact cell pellet after the final spin step (Figure 2A). Once plated these cultures will be relatively free of cellular debris when viewed under a microscope (Figure 2B). A suboptimal isolation may result in a large and fluffy pellet due to the presence of large amounts of cellular debris. This debris will adhere strongly to the PLL-coated vessel and may interfere with the culture (Figure 2C). If a large and fluffy pellet materializes, it may help to resuspend the cells in 10 ml of PBS and repeat the final spin for 10 min at <300 x g. Once the isolation is complete, the purity of the cultures can be assessed by flow cytometry to identify the percentage of A2B5-positive cells. A successful isolation should result in a pool of cells that are >95% positive for A2B5 (Figure 2D–F). Compared to cells stained with a fluorescent dye-conjugated anti-A2B5 antibody, unstained cells should appear as a negative population and can be used as a baseline reference for gating. Moreover, we have previously shown that selection of OPCs using the A2B5 antigen yields cultures that stain positive for PDGFRα by immunocytochemistry11.
As demonstrated by immunocytochemistry, OPCs at the start of treatment (Day 0) are Olig2+/MBP– whereas cells are Olig2+/MBP+ by Day 4 (Figure 3). The optimal confluency of OPCs and oligodendrocytes at Day 0 and Day 4 are shown in representative bright field images (Figure 4A). The absence of MBP expression at Day 0 serves as a baseline for quantifying oligodendrogenesis. Spontaneous differentiation as a result of PDGF-AA withdrawal serves as a negative control, whereas 45 nM thyroid hormone (triiodothyronine; T3) can be used as a positive control11,15. Quetiapine, also known as Seroquel, and clemastine, also known as Tavist, are FDA-approved drugs that have been shown to accelerate in vitro oligodendrogenesis and can be used as additional positive controls16,17. Figure 4B shows a representative two-channel infrared fluorescent scan demonstrating the expected results for each of these conditions in triplicate. Actin and MBP signals were pseudocolored green and red respectively so that highly differentiated cultures appear yellow in merged images. Results show that MBP expression at Day 4 in the PDGF withdrawal condition was 4.5 ± 1.4-fold higher compared to Day 0, whereas the fold changes in MBP due to treatment with thyroid hormone, quetiapine, and clemastine were 33.9 ± 4.1, 33.4 ± 1.0, and 32.8 ± 0.5 respectively (Figure 4C). The robustness and accuracy of the assay are demonstrated by the small standard deviations among the triplicate samples and the large treatment activation window that is about 20 standard deviations above the PDGF withdrawal control.
While Actin serves as a reliable reference gene for normalization in oligodendrocyte cultures, alternative reference genes can also be used. Olig2 is a nuclear transcription factor that is constitutively expressed in cells of the oligodendrocyte lineage. Its specific and constitutive expression pattern thus may provide a more preferable normalization strategy in heterogeneous culture situations. Figure 5 shows that in OPC cultures differentiated with 10 nM thyroid hormone, the calculated fold change in MBP expression relative to PDGF withdrawal is the same when using either Olig2 or Actin for normalization. Presumably, since the OPC cultures in this experiment were very pure, both proteins can be used reliably for normalization. However, we often observe a small population of Olig2-negative cells by Day 4, but the ratio of this population to the Olig2-positive population does not seem to significantly differ between the PDGF withdrawal and T3 treated group. Thus, the fold-changes are not affected. Given a scenario where a treatment might cause proliferation of Olig2-negative cell types, the choice of normalization antibody should be carefully considered.
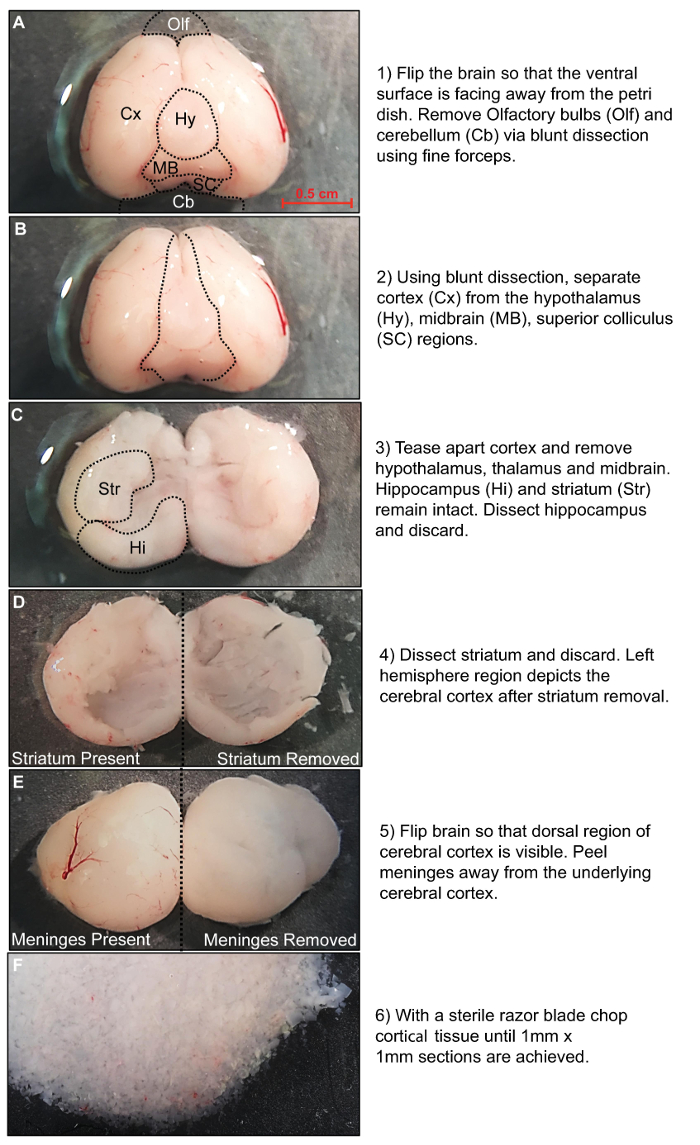
Figure 1: Rat brain dissection procedure. Stepwise description of the dissection procedure. Please click here to view a larger version of this figure.
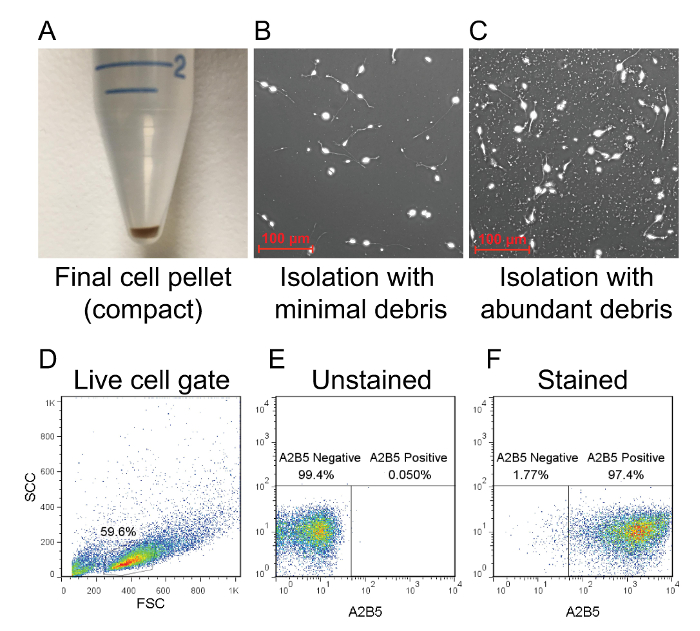
Figure 2: Flow cytometry analysis of isolated OPCs. (A) After magnetic column purification, the bound cell population is plunged out of the column and centrifuged to form a compact pellet. The amount of debris is indicated by the size of the pellet. (B) If the pellet is compact, then debris will be minimal in the culture. (C) A large and fluffy pellet typically results in a culture with heavy debris. The purity of the culture is determined by flow cytometry using a rat anti-mouse A2B5 antibody that is conjugated to APC. (D) Dead cells and debris are excluded from the analysis as shown in forward scatter and side scatter plots. (E) The A2B5 positive population can be determined by using an unstained sampled as a baseline reference for gating. (F) Successfully isolated OPCs should be >95% positive for A2B5. Please click here to view a larger version of this figure.
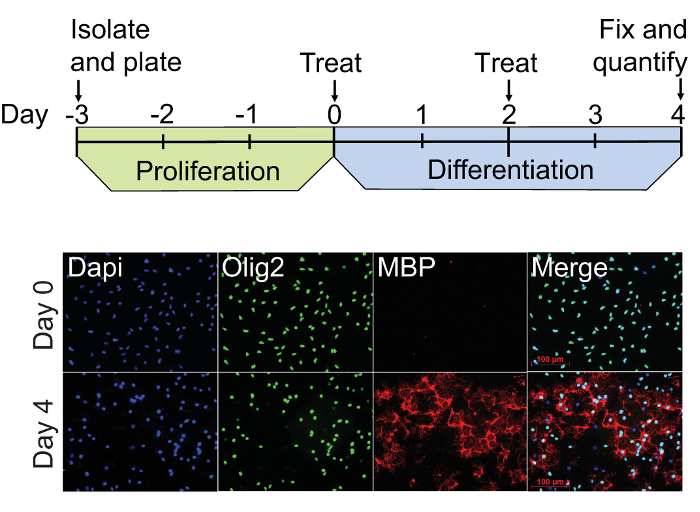
Figure 3: OPC culture and differentiation procedure. The dual-infrared fluorescence-scanning (DIFS) assay begins with freshly isolatedA2B5-positive rat OPCs plated on PLL-coated culture vessels (Day 3). These cultures are proliferated for 3 days in the presence of 20 ng/ml PDGF-AA, and then treated with experimental factors in fresh PDGF-free media on Day 0. Treatment media is fully replenished on Day 2, and the cells are fixed with 4% paraformaldehyde on Day 4. Immunocytochemistry for Olig2 (green) and MBP (red) was performed on representative cultures at Day 0 and Day 4 using Dapi (blue) as a nuclear counter stain. These cultures exhibit constitutive staining for the pan oligodendrocyte lineage cell marker Olig2 at Days 0 and 4, whereas staining for the mature oligodendrocyte marker MBP is only evident by Day 4. Please click here to view a larger version of this figure.
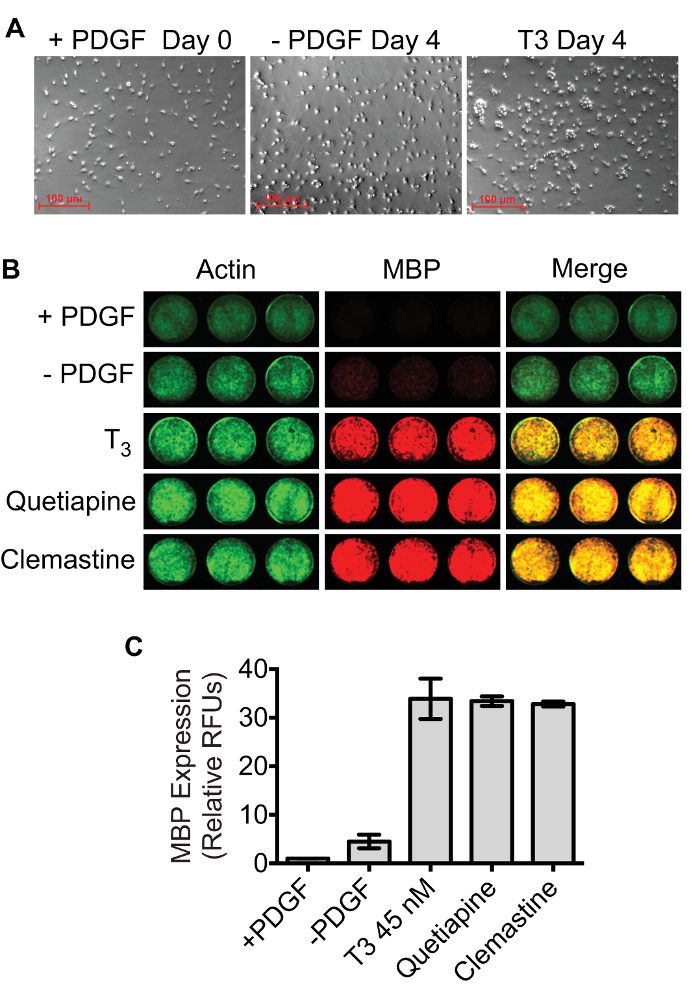
Figure 4: MBP quantification using the dual-infrared fluorescence-scanning (DIFS) assay. (A) The density of OPC cultures at Day 0 and Day 4 required for optimal assay performance. (B) Representative dual-infrared fluorescence scans. OPCs were differentiated in 24 well plates for 4 days in the presence of 45 nM T3, 1 µM quetiapine, 1 µM clemastine, or 0.1% DMSO in triplicate. A separate plate containing OPCs that were fixed on day 0 (+PDGF) was processed in parallel. Actin (pseudocolored green) and MBP (pseudocolored red) were simultaneously quantified using a Licor Odyssey scanner set to detect 700 nm and 800 nm emissions. (C) MBP signals were normalized to Actin signals, and results were scaled to the Day 0 +PDGF control. The mean relative fluorescence units ± SD were plotted using GraphPad Prism 6. Please click here to view a larger version of this figure.
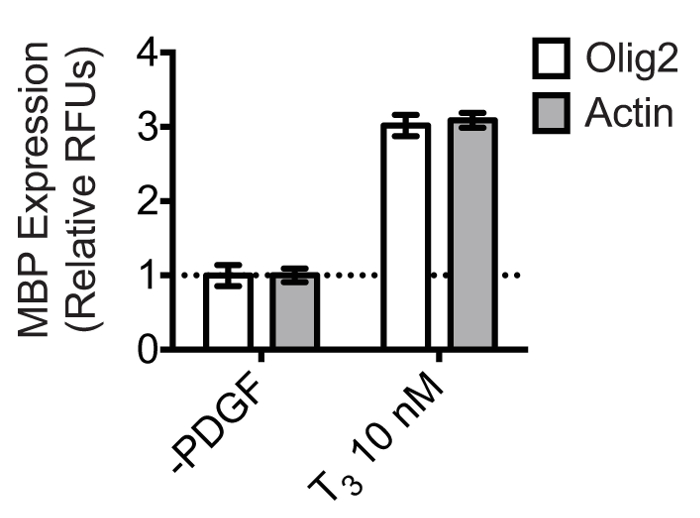
Figure 5: Olig2 is a reliable reference protein for normalizing MBP expression in oligodendrocytes. OPCs were differentiated in triplicate in the presence of 10 nM T3 or 0.1% DMSO vehicle control in 24 well plates for 4 days. The dual-infrared fluorescence-scanning assay was performed using anti-Olig2 or anti-Actin primary antibodies for normalization. Olig2/Actin and MBP were simultaneously quantified using a Licor Odyssey scanner set to detect 700 nm and 800 nm emissions. MBP expression was normalized to either Olig2 or Actin expression, and results were scaled to the 0.1% DMSO control. The mean relative fluorescence units ± SD were plotted using GraphPad Prism 6. Results show that the calculated fold-change is the same when normalizing to either of the two reference proteins.

Figure 6: Effect of Triton X-100 on MBP staining. OPCs were differentiated in 24 well plates for 4 days in the presence of 45 nM T3 or 0.1% DMSO. Cultures were then fixed with 4% PFA and stained for Olig2 and MBP using two different immunocytochemistry protocols. For the high Triton X-100 sample, the cells were permeabilized for 1 hr with 0.4% Triton X -100 and all antibody incubation steps were performed in the presence of 0.1% Triton X-100. For the low Triton X-100 sample, the cells were permeabilized for 1 hr with 0.1% Triton X-100, while antibody incubations were performed without Triton X-100. Olig2 and MBP were visualized with fluorescent dye-conjugated secondaries and images were acquired using a camera-based fluorescence microscope. Please click here to view a larger version of this figure.
