We discovered that neogenin is transiently expressed during the early developmental stages of preimplantation mouse embryos, appearing as early as at the 2-cell stage and lasting until the early morula but becoming deficient at the late morula and blastocyst stages (Figure 2A). In addition, the spatial distribution of neogenin was restricted mainly to outside cells. The results of RT-PCR analysis were consistent with the early and transient nature of neogenin expression, peaking at the 4-cell stage but completely lacking at the 16-cell stage or later (Figure 2B). By contrast, F-actin did not reveal such a transient and polarized expression pattern.
We next investigated whether neogenin levels could be manipulated by microinjection of either neogenin cDNA vectors (neogenin gain) or vectors harboring shRNA targeting neogenin (neogenin loss) into the 2-PN zygote. To visually differentiate neogenin gain from neogenin loss, RFP and GFP were co-expressed as indicators (Figure 3), and the resulting neogenin expression level was confirmed both by immunofluorescence and by immunoblotting (Figure 4).
Figure 5 shows representative images of embryos at each stage after receiving either neogenin shRNA or neogenin cDNA at the 2-PN stage. There were no apparent gross morphological differences between neogenin loss and neogenin gain embryos, and no developmental potential differences at least until the blastocyst stage (Table 2). In fact, both the number of surviving embryos at each stage and the number of arrested embryos were not significantly different.
However, ICM development was less pronounced in neogenin loss than neogenin gain embryos as evidenced by a higher expression of ICM-specific Oct3/4 in neogenin gain embryos (Figure 6A) and by higher numbers of Oct3/4-positive ICM cells per blastocyst in neogenin gain than neogenin loss embryos (Figure 6B). Furthermore, RT-PCR analysis revealed a strong correlation between the expression level of three transcription factors and that of neogenin. In neogenin loss blastocysts, little Oct3/4, Sox2, and Nanog were detected, which is in sharp contrast to a significant increase in the expression of all three transcription factors in neogenin gain over control embryos (Figure 6C). Unexpectedly, the expression levels of Cdx2 and Tead4, transcription factors implicated in TE differentiation/maintenance6,7, were less influenced by the expression level of neogenin, validating that up-regulation of neogenin leads to the activation of transcriptional regulators specific for ICM but not for TE establishment. All data shown are modified from reference9.
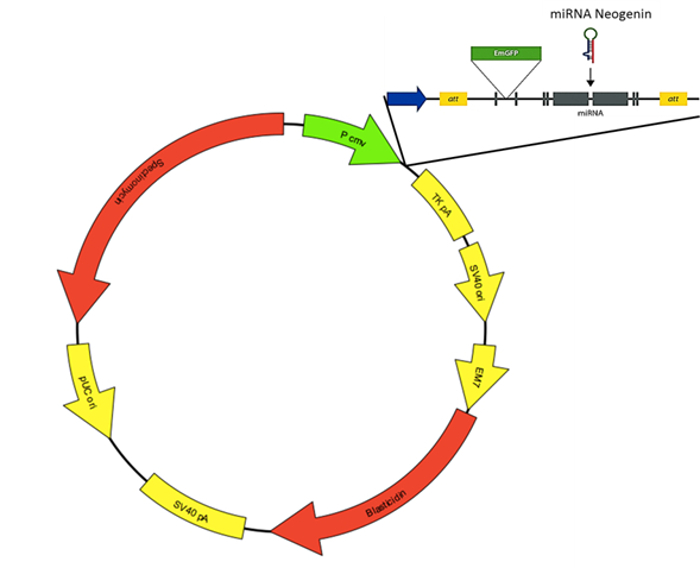
Figure 1: The Structure of pcDNA-EmGFP-miR-neogenin. Please click here to view a larger version of this figure.
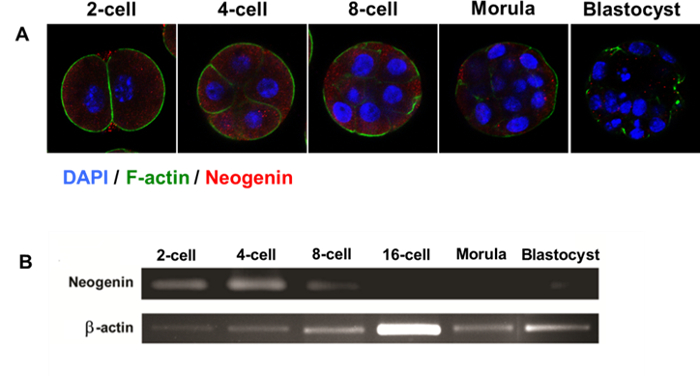
Figure 2: Expression of Neogenin During Mouse Embryonic Development. (A) Confocal microscopic images of neogenin expression in preimplantation embryos at different developmental stages. (B) The reverse transcription polymerase chain reaction of neogenin mRNA in preimplantation embryos at different developmental stages. β-actin was used as an internal control. Please click here to view a larger version of this figure.
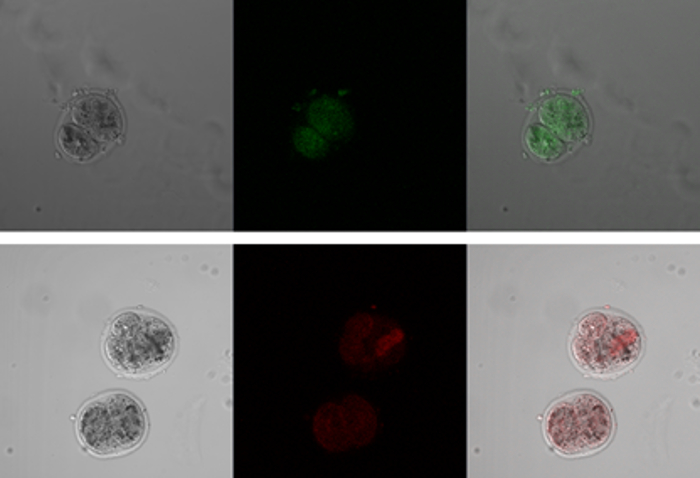
Figure 3: Green Fluorescent Protein (GFP) and Red Fluorescent Protein (RFP) Expression as Indicators for Neogenin Loss and Neogenin Gain, respectively. After microinjection of a neogenin-targeting shRNA vector harboring conjugated GFP or microinjection of a neogenin cDNA vector harboring RFP into 2-PN zygotes, the expression of GFP and RFP was visualized under a fluorescence microscope at the 2-cell and 4-cell stages. Left panel, phase-contrast images; middle and right panels, fluorescence images. Please click here to view a larger version of this figure.
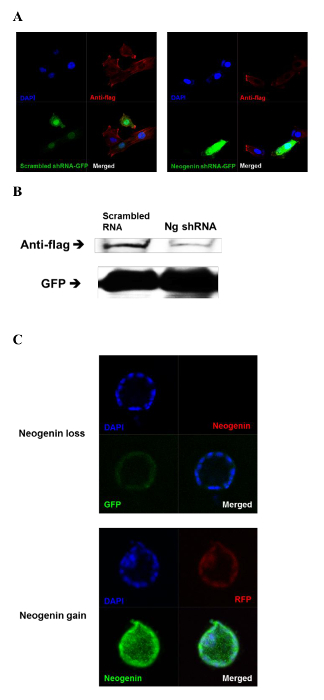
Figure 4: Expression of Neogenin in a Blastocyst After Microinjection of either shRNA Targeting Neogenin or Neogenin cDNA. (A) After microinjection of a neogenin-targeting shRNA vector into 2-PN zygotes, the expression level of neogenin in individual blastocyst cells was evaluated by immunostaining with anti-flag antibodies. In the left panel, scrambled neogenin shRNA vectors were microinjected (control). In the right panel, neogenin-targeting shRNA vectors were microinjected. DAPI, DAPI (blue) was used to stain the nucleus; Anti-flag, visualization of the flag tag on neogenin (red); GFP, green fluorescent protein (green); merged, superimposition of DAPI, anti-flag, and GFP. (B) Whole cell lysates of blastocysts were immunoblotted with anti-flag antibodies. GFP was used as a loading control. Scrambled shRNA, scrambled neogenin shRNA injection; Ng shRNA, neogenin-targeting shRNA injection. (C) The neogenin cDNA vectors or the neogenin-targeting shRNA vectors were microinjected into the 2-PN zygotes and the resulting blastocysts were subjected to immunostaining with anti-neogenin antibodies to measure neogenin expression level. In the upper panel, neogenin-targeting shRNA was injected. Blastocysts were immunostained with anti-neogenin antibodies (red). GFP, green fluorescent protein (green); Merged, superimposition of DAPI, anti-neogenin, and GFP. In the lower panel, neogenin cDNA vectors were microinjected. Blastocysts were immunostained with anti-neogenin antibodies (green). DAPI, DAPI nuclear staining (blue); RFP, red fluorescent protein (red); Merged, superimposition of DAPI, RFP, and anti-neogenin. Please click here to view a larger version of this figure.
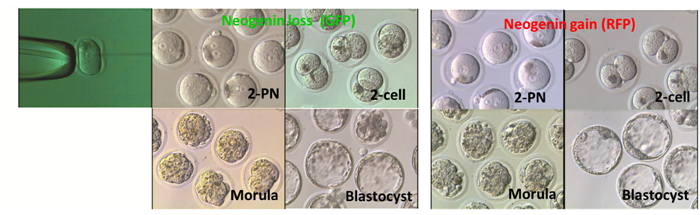
Figure 5: Effects of Neogenin Loss or Gain on Embryonic Development. 2-PN mouse embryos were microinjected either with small hairpin RNA (shRNA) targeting neogenin (neogenin loss) or with vectors harboring neogenin cDNA (neogenin gain) and were cultured until reaching the blastocyst stage. To differentiate neogenin loss from neogenin gain embryos, GFP and RFP were co-expressed, respectively. Phase-contrast images of embryos at different developmental stages are shown. Please click here to view a larger version of this figure.
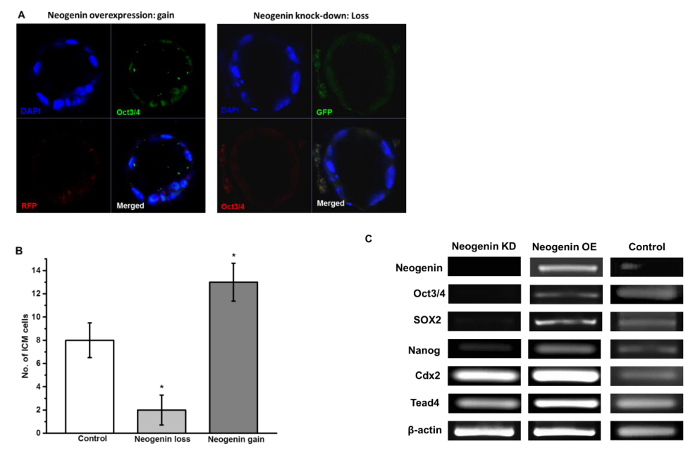
Figure 6: Neogenin Overexpression Favors ICM Differentiation. (A) ICM cells in blastocysts were viewed by immunostaining for Oct3/4. DAPI was used to stain the nucleus. (B) The number of Oct3/4-positive ICM cells in a blastocyst of control, neogenin loss, and neogenin gain embryos are represented as the mean ± SEM from three independent experiments with at least 10 embryos used in each experiment. *Significant differences from control at p <0.05 by Student's t-test. (C) RT-PCR analysis of Oct3/4, Sox2, Nanog, Cdx2, and Tead4 mRNAs from blastocysts of neogenin loss, neogenin gain, and control embryos. β-actin was used as an internal control. Please click here to view a larger version of this figure.

Table 1: Sequences of the Primers and PCR Conditions Used for RT-PCR.
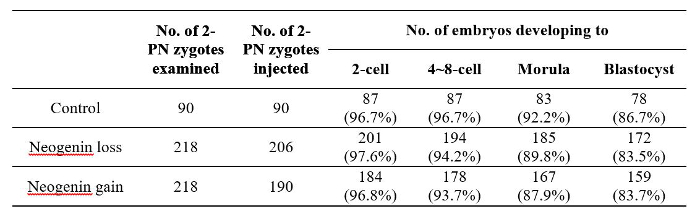
Table 2: The Number of Surviving Mouse Embryos at Each Developmental Stage After Receiving Neogenin shRNA or Neogenin cDNA at the 2-PN Stage.
