As illustrated in Figure 1, sheets of keratocytes can be observed migrating out from beneath the scale within hours of establishing the explant culture. Occasionally, an individually migrating keratocyte which has broken away from the collectively migrating sheet can be observed. In addition, as the explant was established from a fish which had been previously plucked, there are abundant neutrophils migrating through the sheet. At longer culture periods, the keratocyte sheet fragments as cells undergo EMT14.
The initial rate of sheet advancement is extremely rapid but this rate quickly slows as cells spread (measured by area and internuclear distance which increase ~1.8 and ~3.1 fold respectively) during the first 48 hr of culture (Figure 2A-C). The rapid increase is not associated with cell proliferation; after labeling for 24 hr with the fluorescent thymidine analog ethynyl deoxyuridine (EdU), ~10% of cells show evidence of cell division, a rate far lower than seen in transformed cell lines but more consistent with the cells involved in reepithelization in human organotypic cultures30. At later times, the sheet fragments (a phenomenon associated with the EMT observed in this system14) which makes the advance of the leading edge difficult to measure.
The area covered by sheets after 24 hr, can be used as a rapid screen for the ability of compounds to alter collective cell migration15,31. When treatment is added at the time explants are established, a dose-response in sheet area can be seen. Some mammalian cytokines (e.g., TGF-115), chemokines (such as CXCL12) and small molecule inhibitors originally characterized in mammalian systems (e.g., MMP-2, -9,and -13 inhibitors31) have been successfully employed. However, as sheet areas at 24 hr are not normally distributed (Figure 3A), non-parametric statistical tests such as a Kruskal-Wallis ANOVA must be used to determine statistical significance. For graphical display, the median standard errors of the median (upper reference limit (URL) and lower reference limit (LRL)) are plotted as these accurately describe the variation in the data. The URL and LRL are the data points (where n = number of observations) above and below the median in a rank order list of the data. When using custom error bars in Microsoft Excel, the difference between the median and each reference limit must be used (UEB and LEB in Figure 3C).
In many cases, effects of treatment(s) on collective migration are observed in shorter periods of time. In these cases, video microscopy can be used to assay response of the collectively migrating sheet to treatment. When soluble RGD containing peptide is added to the culture medium (third panel in Figure 4) to disrupt adhesion formation, the lamellae at the leading edge of the sheet rapidly shrink and subsequently the leading edge of the sheet detaches and retracts. As the entire sheet retracts in less than 2 min, response of the sheet to treatment can be rapid.
To assess localization of proteins within the sheet or within leader and follower cells, the entire sheet can be fixed and stained, as shown in Figure 5. Care must be taken during the fixation process as the cell sheets are easily disrupted. In our hands, many polyclonal antibodies to the functional domains of mammalian proteins (particularly of cytosolic proteins) have been found to cross react in our zebrafish explants.
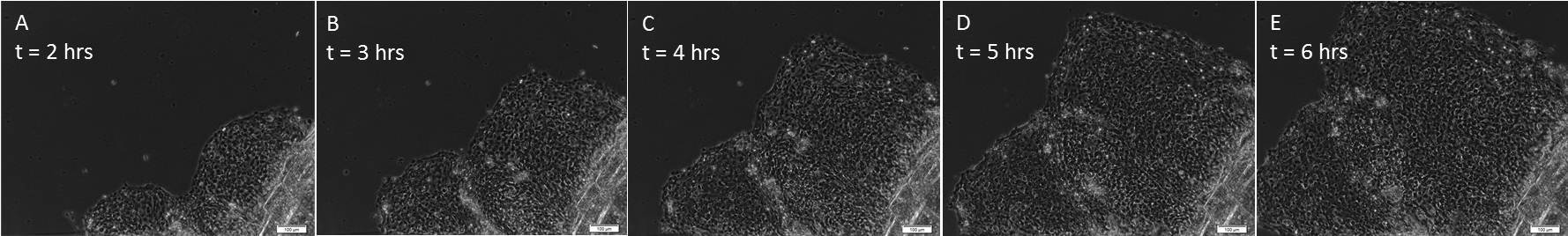
Figure 1. Collective migration of cell sheets. After establishment in explant culture, keratocytes migrate out from underneath the scale as a collective unit. (A) illustrates the relative amount of cells that have migrated away from the scale after only 2 hr in culture, with (B-E) taken each hour thereafter (3 – 6 hr, respectively). All images were taken at 100X magnification. The entire sequence is shown in Video 1; one frame was taken every 30 sec over the 6 hr time period. Please click here to view a larger version of this figure.
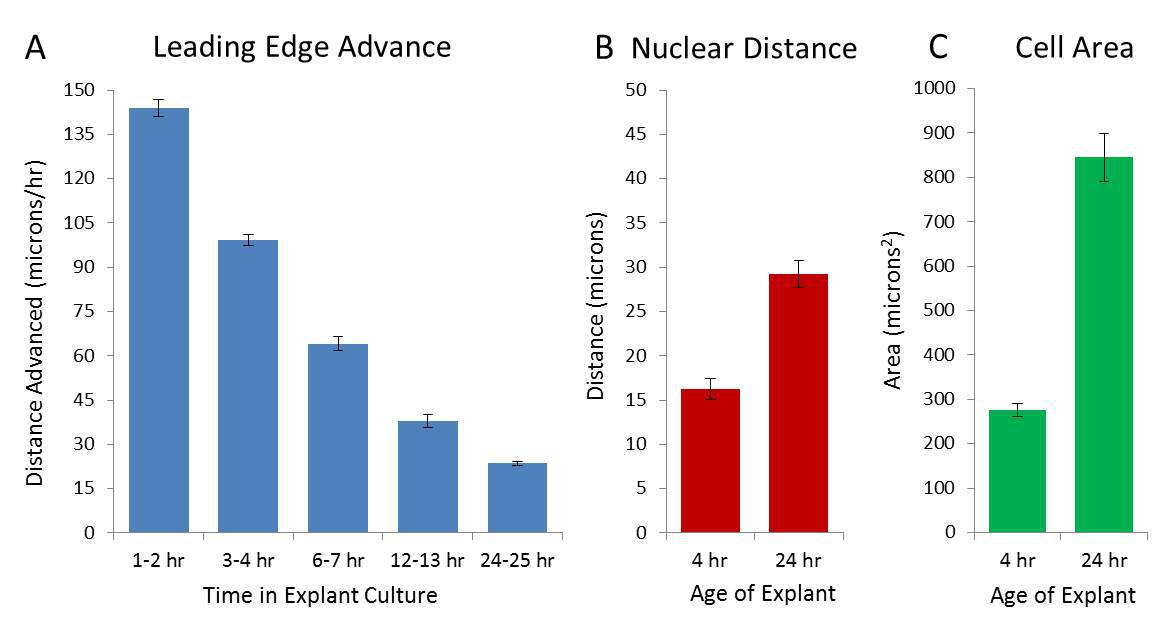
Figure 2. Characteristics of Collective Migration. Initial rate of advance of the leading edge, measured at several points, is rapid but slows down during the first 24 hr in culture (A). Measurement of internuclear distance and cell area using cultures fixed at 4 and 24 hr and stained with DAPI and phalloidin as reference indicate that during the same time period, both the internuclear distance (measured from the center of each nucleus) and the cell area increases (measured as area enclosed by the cortical actin cytoskeleton, (B) and (C) respectively). Each graph represents the mean (± SEM) of three independent experiments in triplicate. This figure has been modified from Rapanan et al.7 Please click here to view a larger version of this figure.
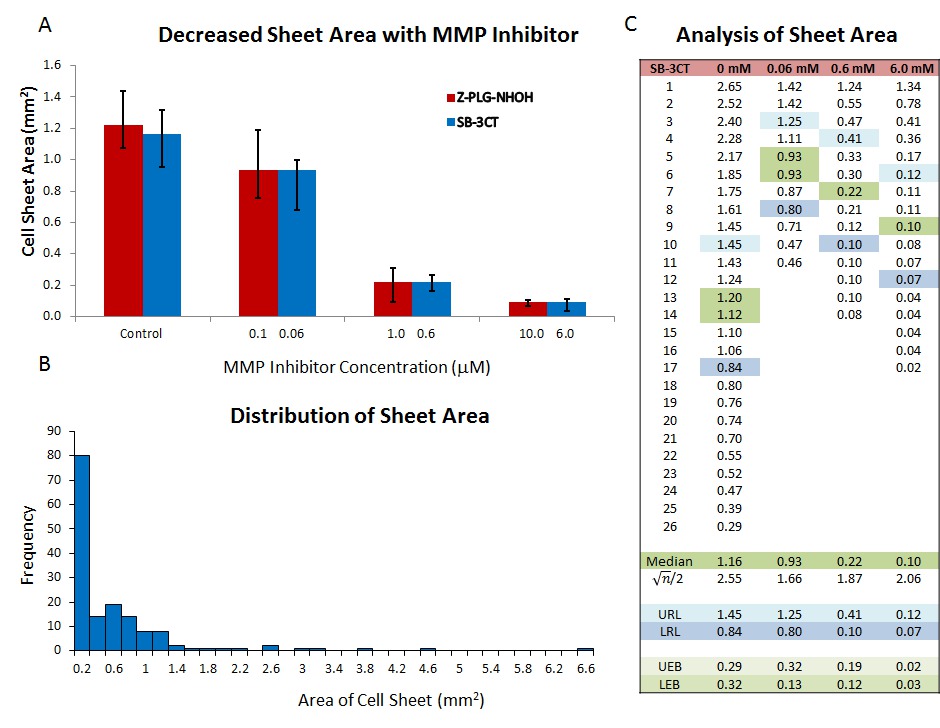
Figure 3. Assay of Collective Migration using Cell Sheet Area. When added to the culture medium during establishment of the culture, peptide Z-PLG-NHOH (broad spectrum MMP) and small molecule SB-3CT (MMP2 & 9 specific) inhibitors decrease cell sheet area after 24 hr in culture (A). As the area of untreated cell sheets is exponentially distributed (B), data should be plotted as medians with standard error of the median determined as described in the text and indicated in blue (upper reference limit or URL and lower reference limit or LRL; (C)). The difference between the median and the URL and LRL determine the size of the error bars on the graph (shaded in light green). This figure has been modified from McDonald et al.31 Please click here to view a larger version of this figure.
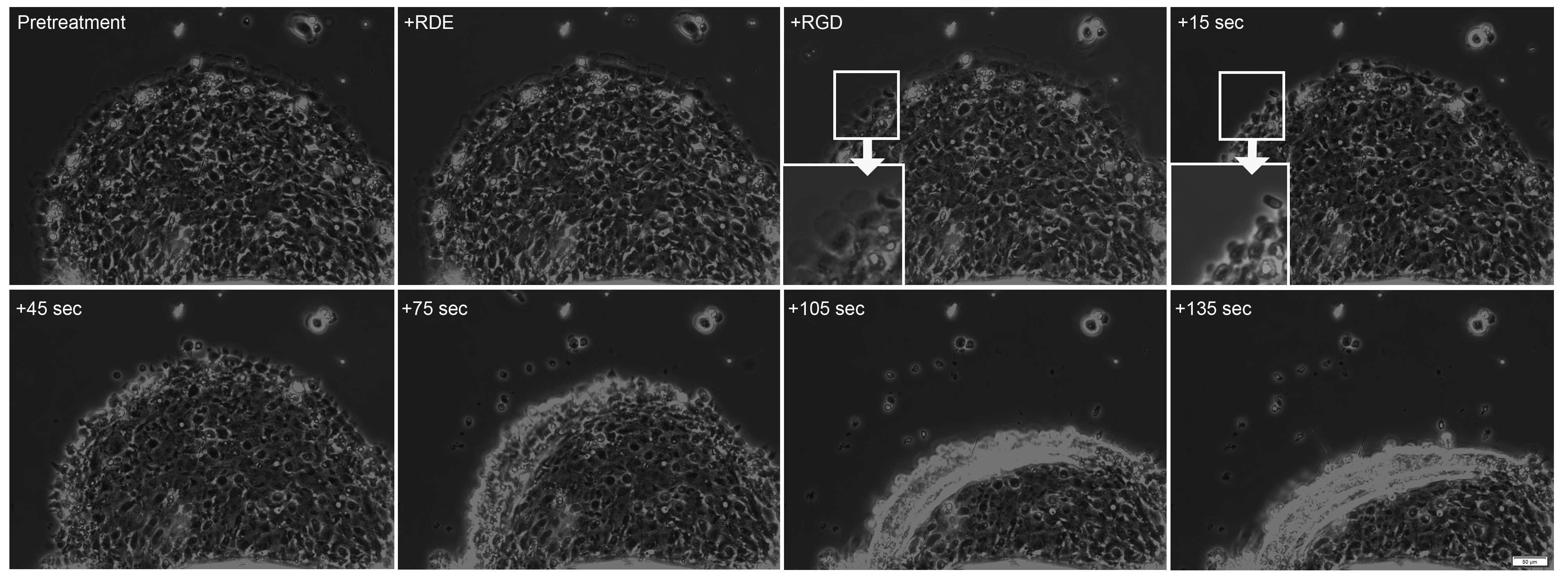
Figure 4. Addition of Soluble RGD Peptide Leads to Sheet Retraction. During pretreatment and after addition of an RGE-containing peptide, the cell sheet continues to advance. 15 sec after addition of an RGD-containing peptide, there is a decrease in the lamellae at the leading edge (inserts). After an additional 30 sec, the sheet begins to retract, a retraction which continues during the duration of the video (total video length ~5 min, see Video 2). Please click here to view a larger version of this figure.
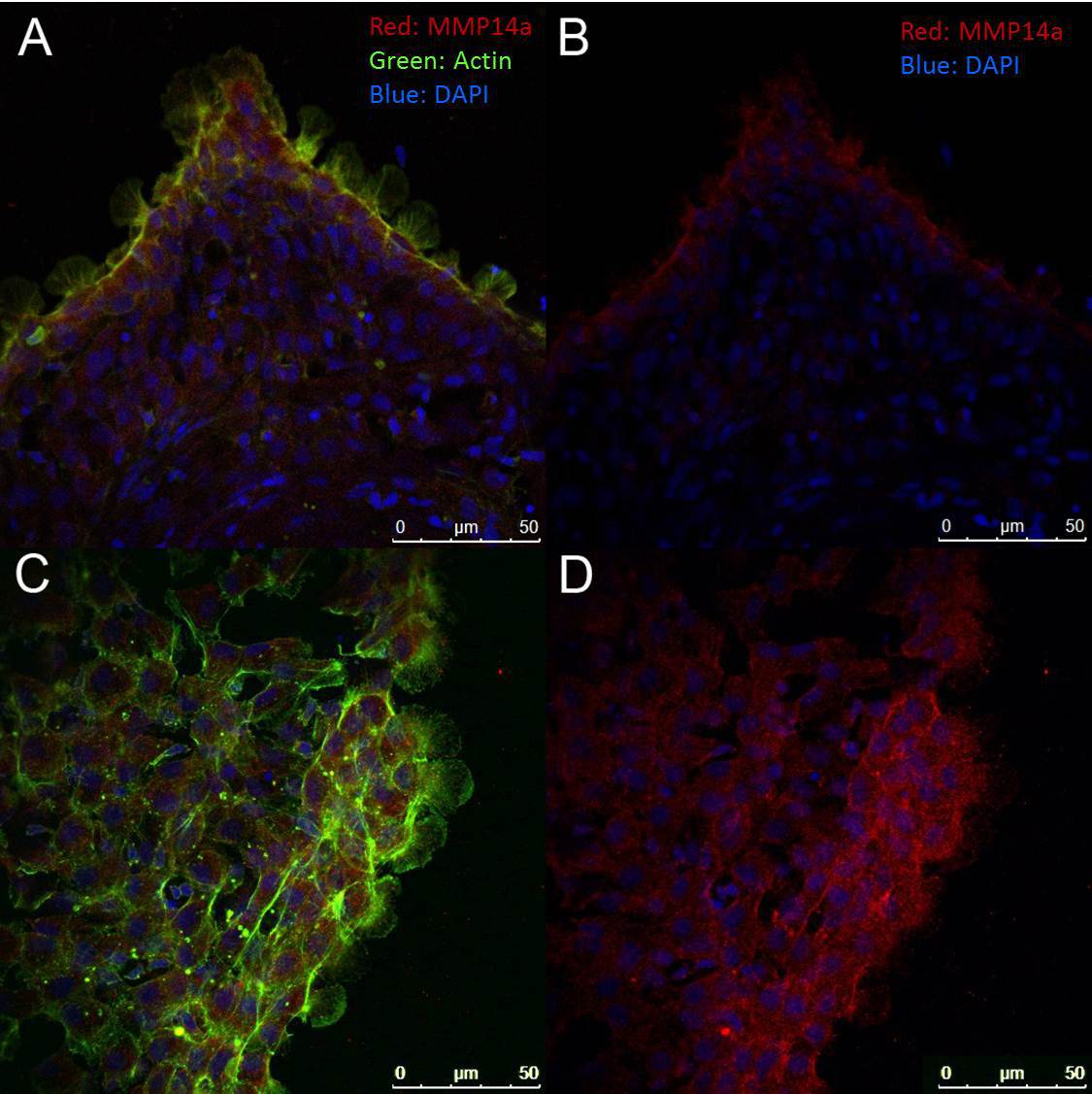
Figure 5. Immunofluorescence Assay. Cells were stained with an antibody to the catalytic domain of MMP14a (red) and counterstained with fluorescently-labeled (488) phalloidin (green) and/or DAPI (blue). (A) and (B) represent a typical 4 hr sheet emerging from the scale while (C) and (D) show a portion of a typical 24 hr sheet. All images were taken at 400X magnification. Note that the intensity of MMP14a staining is higher at the leading edge of the emerging cell sheets (seen in (B) and (D)) and prominent lamellipodia are seen in the leader cells (A) and (C)). Please click here to view a larger version of this figure.
