Variation in plate preparation can greatly influence swarming motility. The curing or drying time after pouring of melted agar medium affects the thin liquid film present on surface motility assays and the bacterial motility over time. Changes in nutrient composition also affect swarming for several bacteria. Figure 1A shows a short-term effect of drying time upon spreading of India Ink and spreading of an initial inoculum of Bacillus subtilis11. Figure 1B shows the effect of drying time and Figure1C shows the effects of ammonium sulfate [(NH4)2SO4] upon subsequent tendril development by swarming P. aeruginosa5.
Quantifiable data can be obtained from endpoint images of surface motility using multiple imaging strategies. Figure 2 shows representative surface growth results for P. aeruginosa swarming and its associated GFP fluorescence image; B. subtilis swarming and its associated bioluminescence image; and Myxococcus xanthus surface growth and the associated red fluorescence image of SYTO 64-stained cells.
Expansion of data acquisition beyond just inspection and imaging of end-point results allows for the study of dynamic behavior(s) for surface growing bacteria. Figure 37 shows an example of P. aeruginosa swarming (imaged for GFP expressing cells) and its associated rhamnolipid production (imaged using Nile Red lipid stain)—the quantification of data from these images is also displayed to show the expansion rate of P. aeruginosa swarming. Video 1 shows a time-lapse of B. subtilis swarming imaged using luminescence of a lux-expressing strain. Video 28 shows a time-lapse of P. aeruginosa (green—expressing GFP) and Salmonella enterica serovar Typhimurium (red—expressing lux) in a competitive swarm assay.
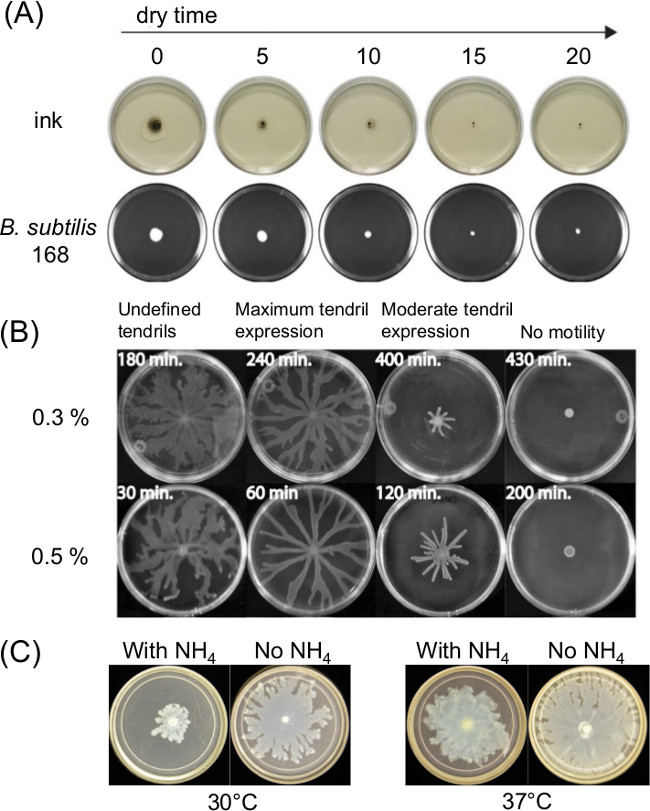
Figure 1: Examples of factors in surface motility assay preparation that affect assay outcome. Effect of (A) agar drying time on agar surface moisture and spreading of inoculum for B. subtilis (Ref8), (B) agar drying time on P. aeruginosa swarming (Reprinted from Ref5 with permission), and (C) presence or absence of ammonium sulfate on P. aeruginosa swarming and tendril formation.
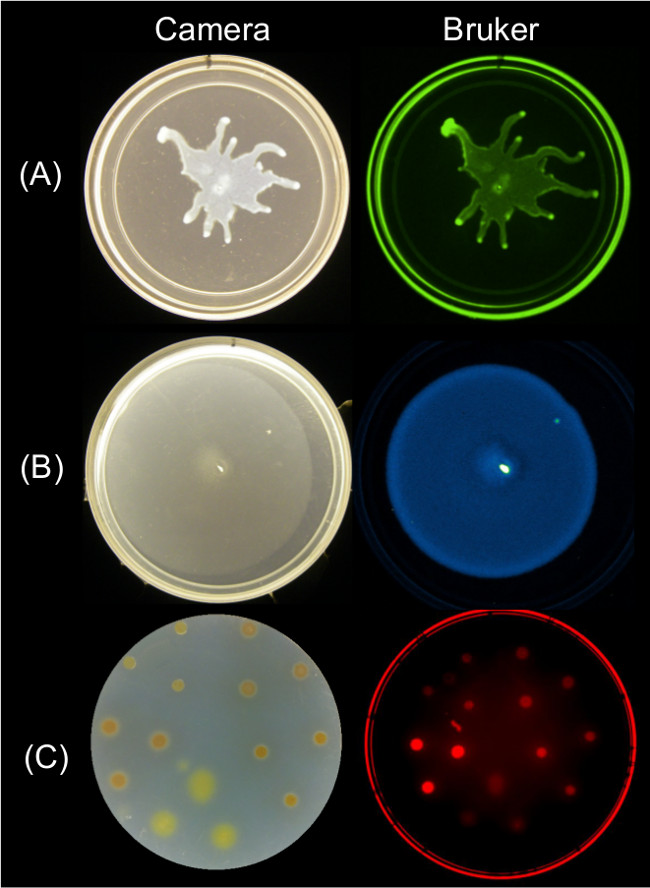
Figure 2: Alternative approaches for imaging surface growth and motility of bacteria using a Bruker imaging station. Side by side image of a camera (left) and Bruker image (right) showing (A) P. aeruginosa expressing GFP—imaged using Green Fluorescence settings, (B) B. subtilis expressing lux bioluminescence reporter—imaged using Luminescence settings, and (C) M. xanthus stained with SYTO 64—imaged using Red Fluorescence II settings. See Table 2 for setting details.
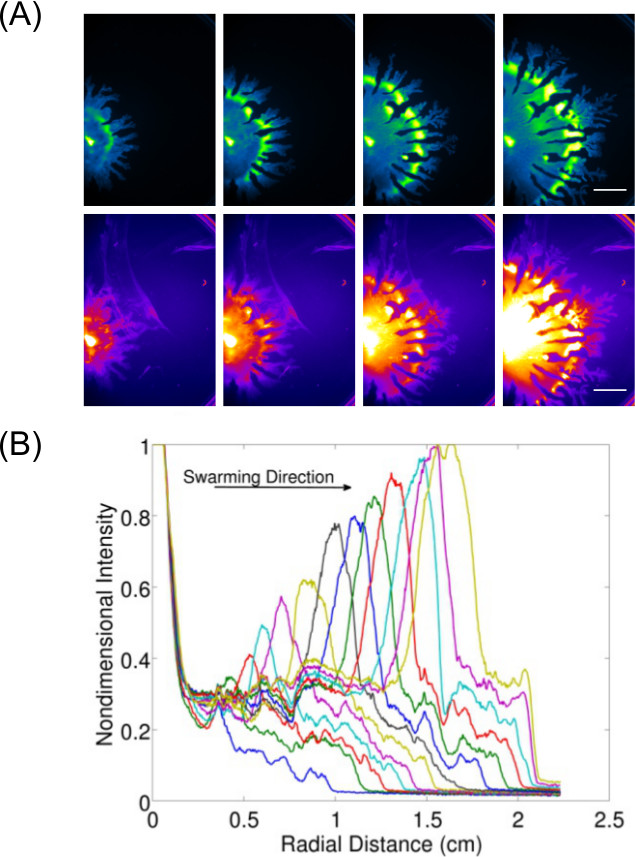
Figure 3: Qualitative and quantitative analysis of a surface motility assay. (A) Time-lapse analysis of cell density distribution, rhamnolipid production (Nile Red lipid stain imaged using the Red Fluorescence I settings; scale bar = 15 mm), and (B) quantification of the of expansion rate from cell density distribution images of a P. aeruginosa swarm. (Reprinted from Ref6 with permission.)
Video 1. Time-lapse imaging of a B. subtilis swarm. B. subtilis expressing lux and recorded using the Luminescence settings. See Table 2 for setting details.
Video 2. Interspecies competition visualized by time-lapse imaging. Swarms of P. aeruginosa (green; expressing GFP and recorded using the Green Fluorescence settings) and S. enterica serovar Typhimurium (red; expressing lux and recorded using the Luminescence settings). See Table 2 for setting details. (Reprint with permission from Ref7.)
| P. aeruginosa | P. aeruginosa tendril formation studies | B. subtilis | M. xanthus | |
| Overnight broth culture media | FAB plus 30 mM Glucose | FAB plus 30 mM Glucose | LB | CTT |
| Overnight broth culture incubation temperature | 37 °C | 37 °C | 37 °C | 30 hr at 30 °C |
| Swarm media | FAB | FAB minus (NH4)2SO4 | 2% (wt/vol) LB | CTT |
| Swarm media: additional components | 12 mM Glucosea | 10% (wt/vol) CAA, 12 mM Glucosea | n/a | SYTO® 64a |
| Agar type | Agar, Noble | Agar, Noble | Granulated agar | Agar, Noble Affymetrix |
| Agar concentration (wt/vol) | 0.45% | 0.45% | 0.60% | 1.50% |
| Swarm plate size | 60 mm | 60 mm | 100 mm | 150 mm |
| Media volume per plate | 7.5 ml | 7.5 ml | Hand Poured | Hand Poured |
| Swarm media setting/drying method | Hood; plates uncovered | Hood; plates uncovered | Benchtop; plates covered | Benchtop; plates covered |
| Swarm media setting/drying time | 30 min | 30 min | Overnight (20 -24 hr) | Overnight (20 -24 hr) |
| Swarm assay incubation temperature | 30 or 37 °C | 30 °C | 37 °C | 30 °C |
| Incubation for time lapse imaging | 30 °C for at least 4 hr | 30 °C for at least 4 hr | 37 °C for 2 hr | RT for 12 hr |
| Time-lapse capture length | 24 hr | 24 hr | 10 hr | 66 hr |
| Time-lapse setting | 1 frame/10 min | 1 frame/10 min | 1 frame/6 min | 1 frame/10 min |
| aAdded after autoclaving. | ||||
Table 1: Specifications for Surface Motility Assay Preparation. Includes surface motility assay preparation specifications for P. aeruginosa, B. subtilis, and M. xanthus.
| Signal | Green Fluorescence | Red Fluorescence I | Red Fluorescence II | Luminescence |
| Protein or dye | Green Fluorescence Protein (GFP) | mCherry protein or Nile Red rhamnolipid stain | SYTO® 64 | Luciferase from lux operon |
| Excitation wavelength (nm) | 480 ± 10 | 540 ± 10 | 590 ± 10 | Off |
| Emission wavelength (nm) | 535 ± 17.5 | 600 ± 17.5 | 670 ± 17.5 | No filter |
| Exposure time (sec) | 30 | 60 | 60 | 240 |
| f-stop | 4.0 | 4.0 | 2.5 | 1.1 |
| FOV (mm) | 190 | 190 | 140 | 120 |
| Focal plane (mm) | 27.5 | 27.5 | 12.2 | 4 |
| Binning (pixels) | None | 2 x 2 | None | 8 x 8 |
Table 2: Imaging Specification. Bruker imaging station specifications for red and green fluorescence, and luminescence imaging of bacterial surface growth.
