Thawing cells
- Obtain frozen mouse embryonic fibroblasts (MEFs) (generated previously from individual embryo primary cultures) from liquid nitrogen.
- Thaw cells by immersing vial in 37°C water with swirling until contents are completely thawed.
- Add 9mL of DMEM that has been supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin to a 10cm cell culture plate.
- Add the thawed cell suspension drop-wise to the plate, rock the plate gently to disperse cells and place plate in a 37°C, 5% CO2 incubator overnight. Note that if cells are sensitive to DMSO (from the freezing process), initial media can be replaced with fresh media after cells have adhered to the plate (approximately four hours post plating). Also note that confluence of the cells after thawing depends on the number of cells frozen, the recovery rate of the cells after freezing, and the growth rate of the cells.
Passaging cells
- When cells have grown to essentially 100% confluency (ie: the next day), aspirate off the media and wash cells with 5mL of sterile PBS.
- Remove PBS and add 1mL of trypsin containing 10mM EDTA and place plate in incubator for approximately 5 minutes to allow cells to detach from plate.
- When cells have detached, add 9mL of media to the trypsinized cells, pipette 2-3 times to disperse clumps, add 1mL of this suspension drop-wise onto a new plate containing 9mL of media, and place this into the 37°C incubator overnight. The presence of FBS in the media will inactivate the trypsin – alternatively, cells can be washed first to dilute out the trypsin.
Counting and plating cells onto cover slips
- Remove cells from incubator, wash and trypsinize as before, except this time add 5-6mL media back to the trypsinized cells instead of 9mL.
- Pipette cells thoroughly to ensure complete dissociation into a single cell suspension, and remove 15μL to combine with 15μL of trypan blue in a separate microfuge tube.
- Mix this cell/ trypan blue suspension by pipetting up and down several times, and add 15μL to the hemocytometer.
- Under the microscope, dead cells will appear blue. Count the number of clear/colourless (living) cells in one of the 4X4 grids on the hemocytometer. Repeat this count on the three other 4X4 grids (denoted A, B, C and D) and use the following equation to calculate the number of cells per μL of suspension:
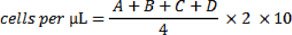
- Through protocol optimization, we have determined that plating 30,000 cells results in appropriate cell density for visualization at the conclusion of the experiment.
- To calculate the volume of cell suspension needed for 30,000 cells, use the following equation:
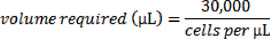
- Now place a sterile glass cover slip into each well of a six well dish and add 2mL of media to each well, and then add drop-wise from your cell suspension the volume required for 30,000 cells into each well. Note that glass cover slips are used for this experiment as we will be visualizing the cells using fluorescence microscopy at a later time point; not all applications will necessarily require microscopy, and thus cover slips may not be required.
Virus Transduction
- After several hours or overnight (sufficient time to ensure that the cells adhere to the cover slips), remove media and wash cells with 1mL of serum-free DMEM.
- Add 1mL of serum-free media to each well for the addition of the virus. For this experiment, adeno-Cre virus was obtained commercially from Vector Biolabs.
- Previous protocol optimization has shown that a multiplicity of infection (MOI) of 500 provides highly efficient gene transduction into primary MEFs with little or no effect on cell viability. MOI can be calculated as follows:

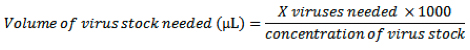
- Add the calculated volume of virus directly to each well that is to be infected, gently rock the plate to disperse the virus and place cells into the 37°C incubator overnight.
- The next morning, remove the virus-containing media from the cells and wash cells in 1mL sterile PBS and then add 2mL of regular media to each well.
- In the case of the Rac1 cells exemplified in this experiment, morphological changes occurred in the cells at approximately 72 hours post transduction.
Representative Results
Cells were visualised by staining with the actin dye phalloidin (for imaging purposes only) and imaged using the Leica TCS-SP5 multiphoton confocal laser scanning microscope at the Advanced Analysis Centre at the University of Guelph. Figure 1 shows the contracted and elongated morphology of the Rac1flox/flox cells that were exposed to adeno-Cre virus compared to the same cells not exposed to virus.
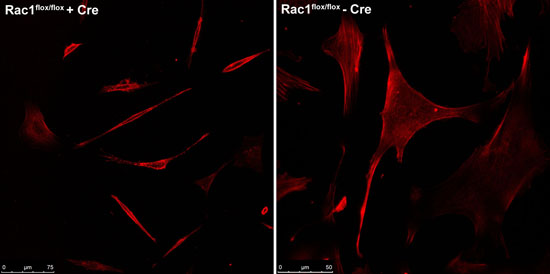
Figure 1. Comparison between Rac1flox/flox cells infected with adeno-Cre virus (left) compared to uninfected cells from the same source (right).
