Generally, designing a DN mutation requires a considerable amount of information on the structure and function of the POI. In contrast, the DN strategy presented here is particularly useful when the structural and functional information for the POI is limited. If the POI is a multimeric protein, a fusion of one subunit to a lysosomal protease can dominantly inhibit the assembled multimer and, potentially, other ligands through a combination of proteolysis of the endogenous subunits and subcellular diversion of the multimer to the lysosome. To confirm successful cloning and test the effectiveness of Dox-regulated transgene activation, purified pTet-Splice plasmid containing the TetO-DN-CB-myc6-Rb1 construct was transfected into mouse NIH3T3 cells that stably expressed the rtTA protein, but not Rb113. In the absence of Dox, cells expressing rtTA displayed no RB1 expression, whereas a robust RB1 expression was observed in the presence of Dox (Figure 2A). Next, to test the reversibility of the system, we cotransfected HEK293 cells (which endogenously express Rb114) with purified pTet-Splice/TetO-DN-CB-myc6-Rb1 and the pCMV-Tet3G vectors. As expected, the expression of the endogenous RB1 protein was significantly inhibited on transgene activation by the addition of Dox in the cell culture media (Figure 2B). To test the reversibility of the system, following a 24 h period, in a subset of HEK293 cells, we replaced the Dox-containing cell culture media with fresh Dox-free media and incubated the cells for an additional 24 h. Supporting the Dox-regulated reversibility, RB1 expression was restored upon Dox removal from the culture media (Figure 2B).Transfected HEK293 cells that were not treated with Dox or Dox-treated HEK293 cells transfected with the pCMV-Tet3G vector only (Figure 2B) showed basal levels of endogenous RB1 expression. Because NIH3T3 cells do not express RB1 protein naturally, the positive RB1 reactivity is due to the accumulation of the transgenic RB1 protein. Given our interest in the potential proliferative effect of the TetO-DN-CB-myc6-Rb1 construct in the auditory system, we measured the total DNA content in pTet-Splice/TetO-DN-CB-myc6-Rb1– and pCMV-Tet3G-transfected HEI-OC1 cells (inner ear organ of a Corti-derived cell line) in the presence and absence of Dox. Consistent with the working hypothesis presented here, a significant increase in the DNA content (corresponding to an increase in cell number) was observed in Dox-treated but not untreated (control) transfected HEI-OC1 cells (Figure 3A – 3C).
Following the in vitro confirmation of the transgene activity and function, a transgenic mouse model was generated. FISH, using a TetO-DN-CB-myc6-Rb1 digoxigenin (DIG)-horse radish peroxidase (HRP)/tyramine-biotin/avidin-Cy5-labeled probe, and G-banding of male mice splenocytes and ear fibroblasts were performed to confirm the transgene insertion in the transgenic mice genome. Among the five germline transmitting founders, line 14 showed a consistent transgene insertion within segment 10C3~D2 (Figure 4A – 4C). Mice from this line were used to establish the breeding colonies and generate experimental animals for further studies. To determine the optimum time for the Dox induction and TetO-DN-CB-myc6-Rb1 transgene activation, we divided the transgenic mice into three different groups, wherein Dox was administered in drinking water for variable lengths of time: Group 1 (3 days of treatment), Group 2 (7 days of treatment), and Group 3 (10 days of treatment). Upon completion of the treatment, changes in the RB1 protein expression were assessed by western blotting (Figure 5A – 5D). Because the DN and native RB1 proteins have similar molecular weights, they cannot be resolved from each other on a western blot. However, consistent with an initial increase in RB1 protein (due to the accumulation of DN-RB1 and endogenous proteins), there was an initial increase in total RB1 protein expression in the transgenic mouse cochleae compared to the control group (Figure 5A). Moreover, consistent with the inhibitory and proteolytic activity of the TetO-DN-CB-myc6-Rb1 transgene product, a quantifiable reduction in RB1 expression was observed in groups 2 and 3 compared to group 1 and control groups (Figure 5A – 5D). Of note, treatment longer than 10 days did not result in any further change in RB1 protein expression. Thus, 10 days of Dox treatment was considered optimal and used for further studies.
Because the rtTA and, consequently, TetO-DN-CB-myc6-Rb1 transgene activation are under the ubiquitous ROSA-CAG promoter, RB1 could be potentially inhibited in any tissue or cell where RB1is endogenously expressed (e.g., lungs, heart, retina). To test this premise and evaluate the transgene's tissue specificity, cochleae, lungs, heart, and eye biopsies were dissected from TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA, and control mice (not carrying the ROSA-CAG-rtTA transgene) were assessed for RB1 expression (Figure 6A – 6C). Regardless of the tissue analyzed, a significant reduction in RB1 protein was observed in TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA but not in the control group (Figure 6A). In sharp contrast, qRT-PCR analyses in the eye, heart, and cochleae of Dox-treated TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA and age-matched control mice showed a significant upregulation of the DN-CBRb transcript in the tissues of the former but not the latter mice (Figure 6C). Altogether, these results confirm the efficiency of the construct. An increase in transcript expression reflects the effective Dox-regulated transgene induction. Likewise, a reduction in protein expression is consistent with the routing and proteolytic degradation of the endogenous RB1.
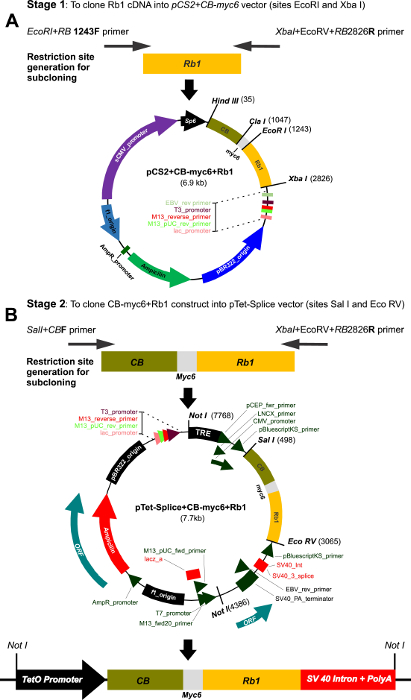
Figure 1: Multi-step cloning of the CBRb and generation of the inducible TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA. (A) This panel shows the cloning of the Rb1 fragment into the pCS2-CB-Myc6 vector. A 1583-bp Rb1 gene product was PCR amplified using the EcoRI+RB1243 forward and Xba+EcoRV+RB2826 reverse primers. The resulting Rb1 fragment was cloned between the EcoRI and Xbal restriction sites of the pCS2 vector containing the CB-Myc6 construct to generate the pCS2+CB-myc6+Rb1 vector. (B) The CB-myc6+Rb1 fragment was cloned into the pTetSplice vector between the SalI and EcoRV restriction sites. Please click here to view a larger version of this figure.
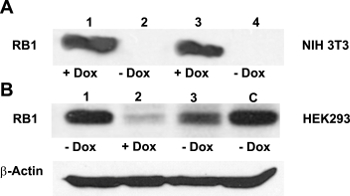
Figure 2: Effectiveness of the Dox-regulated transgene activation. (A) NIH3T3 cells do not usually express RB113. Confirming the efficient activation of the TetO-DN-CB-myc6-Rb1 construct, a western blot analysis with the anti-RB1 antibody revealed a robust RB1 expression in the presence of Dox (lanes 1 and 3), but not in its absence (lanes 2 and 4). (B) HEK293 cells, which endogenously express Rb114, were cotransfected with TetO-DN-CB-myc6-Rb1 and the pCMV-Tet3G transactivator vector. In the absence of Dox (lane 1), a positive RB1 expression was observed. The addition of Dox to the system caused the TetO-DN-CB-myc6-Rb1 activation and RB1 downregulation (lane 2). The RB1 expression was resumed 24 h after Dox was removed from the media (lane 3). C = control, untransfected HEK293 cells not treated with Dox. This is an adaptation from a figure originally published in the journal Frontiers in Cellular Neuroscience2. Its reproduction agrees with Frontiers' policy on authors' copyright retention. Please click here to view a larger version of this figure.
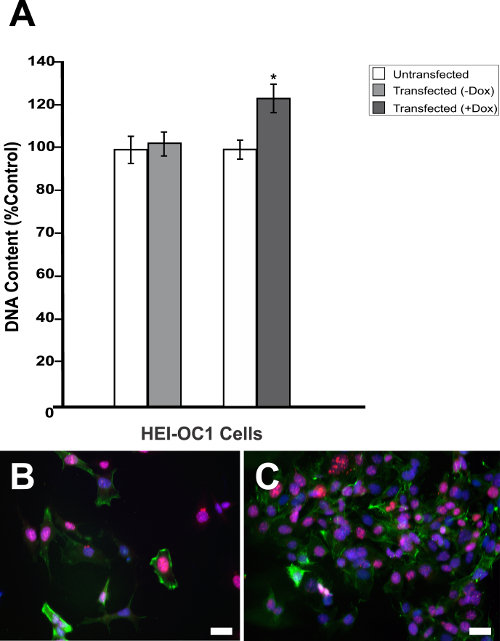
Figure 3: Dox-mediated DN-CBRb transgene activation increases cell proliferation. (A) HEI-OC1 cells cotransfected with purified TetO-DN-CB-myc6-Rb1, and the pCMV-Tet3G vectors were cultured in the absence (−) or presence (+) of Dox. Cell proliferation was determined 48 h after the transfection using a proliferation assay. The percentage change in proliferation was calculated using a change in fluorescence values with untransfected cells as a control. A modest but significant increase in cell number was observed in the transfected cells, following Dox treatment. In contrast, no significant changes were observed between transfected HEI-OC1 cells not treated with (−) Dox and the untransfected control. (B) HEI-OC1 cells untransfected or (C) cotransfected with purified TetO-DN-CB-myc6-Rb1 and the pCMV-Tet3G vectors cultured in the presence (+) of Dox were labeled with Ki-67 (red), Phalloidin (green), and DAPI (blue). * P < 0.05. Scale bar = 10 µm. This is an adaptation from a figure originally published in the journal Frontiers in Cellular Neuroscience2. Its reproduction agrees with Frontiers' policy on authors' copyright retention. Please click here to view a larger version of this figure.
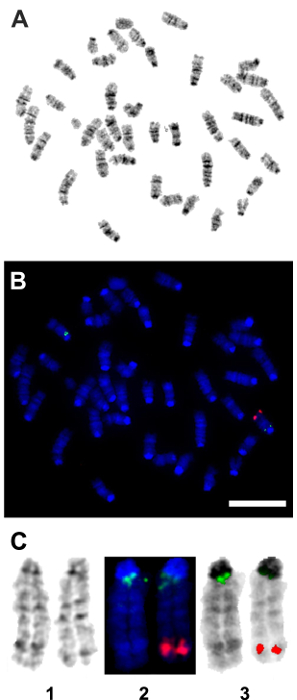
Figure 4: Fluorescent in situ hybridization (FISH) confirmation of the genomic insertion of the TetO-DN-CB-myc6-Rb1 transgene. A pCS2-CBRb (digoxigenin probe/anti-DIG-HRP/tyramide-biotin/avidin-Cy5, in red) test probe was hybridized to splenocytes and ear fibroblast cell metaphase to see if the probe signal could be detected or not with a one-copy target insert size of 2.5 kb. The pCS2-CBRb test probe hybridized to one chromosome 10 with bright, doublet probe signals that showed the insertion of the TetO-DN-CB-myc6-Rb1 transgene within the segment 10C3-D2. The RP23-267G24 (Spectrum Green) control probe hybridized to chromosome 10 at band 10A1 as expected and confirmed that the CBRb DNA of interest inserted into chromosome 10. (A) This panel shows the G-band metaphase. (B) This panel shows the tyramide signal amplification (TSA) FISH image from the same metaphase as shown in panel A. (C) This panel shows composite images of chromosome 10 showing (1) G-banding, (2) TSA FISH, and (3) TSA FISH with inverted DAPI banding. Red = pCS2-CBRb probe; blue = DAPI banding; green = RP23-267G24 control probe. Scale bar = 10 µm. Please click here to view a larger version of this figure.
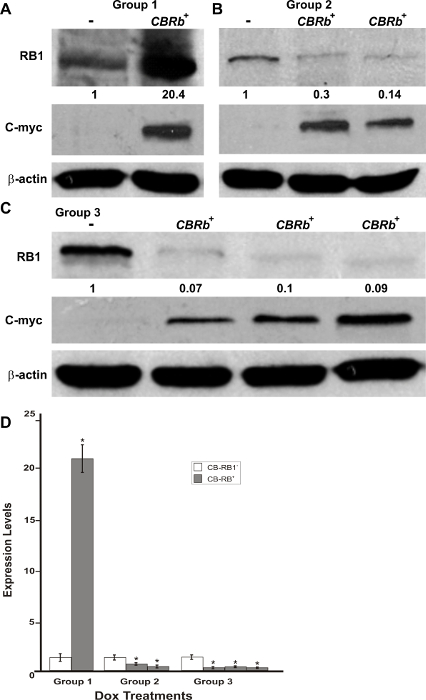
Figure 5: DN-CBRb transgene activation in the inner ear of postnatal (P) 36 TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA+ (CBRb+) and ROSA-CAG-rtTA+ (CBRb−) mice after 3 (group 1), 7 (group 2), and 10 (group 3) days of Dox treatment. (A–C) Anti-RB1, which reacts with both hyperphosphorylated and hypophosphorylated forms of RB1, as well as anti-c-myc antibodies were used for detection. Densitometric analysis was done using ImageJ software. The corresponding values obtained were normalized to the negative control CBRb− (-) and then to β-actin. The relative RB1 expression levels are shown underneath each blot. (D) The graphic representation of normalized RB1 expression levels plotted against the different treatments highlights a consistently lower RB1 detection in CBRb+ (+) samples. Each lane on the western blot gel and each column on the graphic correspond to a different individual. * P < 0.05.This figure was originally published in the journal Frontiers in Cellular Neuroscience2. Its reproduction agrees with Frontiers' policy on authors' copyright retention. Please click here to view a larger version of this figure.
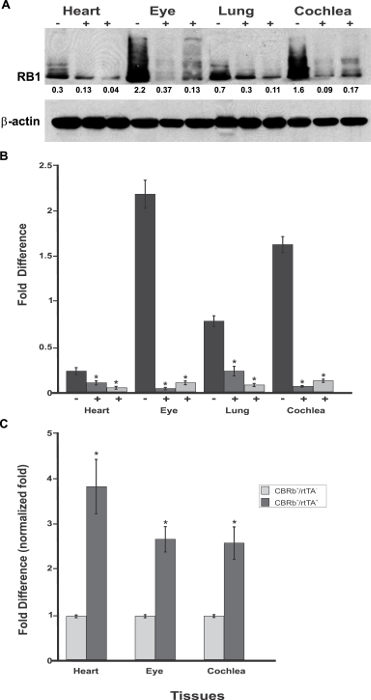
Figure 6: Spatial analyses of the TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA transgene activation in P18 TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA+ (+) and DN-CBRb+/ROSA-CAG-rtTA− negative control (-) mice heart, eye, lungs, and cochlea. (A)Confirming the efficiency of Dox-mediated transgene activation, the expression of the endogenous RB1 was downregulated in the CBRb+/ROSA-CAG-rtTA+ tissues but not in the negative control mice tissues. Densitometric analysis was performed as described in Figure 5. The relative RB1 expression levels are shown underneath each blot. (B) This panel shows a graphic representation of normalized RB1 expression levels plotted against the different tissues. An interindividual variation on endogenous RB1 levels may explain differences in individual responses to transgene activation and RB1 downregulation. (C) RT-PCR analyses in eye, heart, and cochleae revealed a significant upregulation of the TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA transcript in Dox-treated TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA+ tissues, but not in the DN-CBRb+/ROSA-CAG-rtTA− negative control group. CBRb+/rtTA+ = TetO-DN-CB-myc6-Rb1/ROSA-CAG-rtTA+; CBRb−/rtTA+ = ROSA-CAG-rtTA+; * P < 0.05. This figure was originally published in the journal Frontiers in Cellular Neuroscience2. Its reproduction agrees with Frontiers' policy on authors' copyright retention. Please click here to view a larger version of this figure.

Table 1: PCR condition used for checking the CB-Rb1 fusion region.
