Genetic characterization of the latent HIV-1 reservoir, which persists in individuals on long-term antiretroviral therapy (ART), has been vital to understanding that the majority of integrated proviruses are defective and replication-incompetent1,2. During the process of reverse transcription, errors are introduced into the integrated proviral sequence. Some mechanisms that generate defective proviral sequences include the error-prone HIV-1 reverse transcriptase enzyme3, template switching4, and/or APOBEC-induced hypermutation5,6. Two recent studies have found that approximately 5% of HIV-1 proviruses isolated from individuals on long-term ART are genetically intact, and potentially replication-competent, and may contribute to the rapid rebound in HIV-1 plasma levels upon cessation of ART1,2,7. Previous studies have identified that replication-competent HIV-1 proviruses persist in naïve and resting memory CD4+ T cell subsets (including central, transitional and effector memory T cells), indicating the importance of targeting these cells in future eradication strategies2,8,9.
Early insights into the distribution, dynamics and maintenance of the latent HIV-1 reservoir were achieved through utilization of single-proviral sequencing (SPS) methods that genetically characterize sub-genomic regions of the HIV-1 genome10,11,12,13. SPS is a versatile tool, able to sequence a single HIV-1 provirus from within a single infected cell. However, SPS is unable to determine the replication-competency of proviruses, since it only sequences sub-genomic regions and misses proviruses that contain large deletions within primer binding sites. A previous study has demonstrated that SPS overestimates the size of the replication-competent reservoir by 13- to 17-fold through selectively sequencing intact sub-genomic regions2.
To address the limitations of SPS, Ho et al.4 and Bruner et al.1 developed assays to sequence near full-length HIV-1 proviruses. This allowed the frequency of genetically intact, and potentially replication-competent, HIV-1 proviruses in individuals on long-term ART to be determined. These assays amplified and sequenced (via Sanger sequencing) sub-genomic regions that were then assembled to obtain a sequence of the (intact or defective) HIV-1 provirus. Three limitations of this approach are: 1) the use of multiple sequencing primers increases the risk of unintentionally introducing defects into the proviral sequence, 2) primer mismatches may prevent amplification of particular proviruses, and 3) often the entire proviral sequence cannot be resolved due to the technicality of these methods.
To overcome the limitations of existing full-length HIV-1 proviral sequencing assays, we developed the Full-Length Individual Proviral Sequencing (FLIPS) assay. FLIPS is a next-generation sequencing (NGS)-based assay which amplifies and sequences near full-length (intact or defective) HIV-1 proviruses in a high-throughput and efficient manner. FLIPS provides advantages over previous assays, as it limits the number of primers utilized; therefore, it decreases the chance of primer mismatches, which may limit the population of proviruses captured or unintentionally introduce defects into a viral sequence. FLIPS is also less technically challenging than previous assays and involves 6 main steps: 1) lysis of HIV-1 infected cells, 2) amplification of single HIV-1 proviruses via nested PCR performed at limiting dilution using primers specific for the highly conserved HIV-1 5' and 3' U5 LTR region (Figure 1A), 3) purification and quantification of amplified products, 4) library preparation of amplified proviruses for NGS, 5) NGS, and 6) de novo assembly of sequenced proviruses to obtain contigs of each individual provirus.
Sequences generated by FLIPS can undergo a stringent process of elimination to identify those which are genetically intact and potentially replication-competent (Figure 1C)2. Genetically intact proviruses lack all known defects which result in generation of a replication-incompetent provirus. These defects include: inversion sequences, large internal deletions, hypermutation/deleterious stop codons, frameshifts, or mutations in the 5' packaging signal or major splice donor (MSD) site.
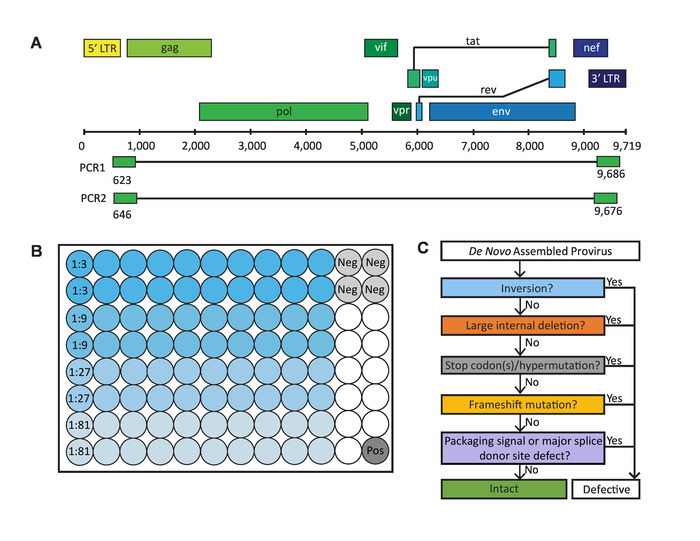
Figure 1: Critical steps in the full-length individual proviral sequencing (FLIPS) assay. (A) HIV-1 DNA genome with primer binding sites in 5' and 3' U5 LTR regions used by FLIPS to amplify near full-length (defective and intact) HIV-1 proviruses via nested PCR. (B) Layout of a 96-well PCR plate containing 80 sample wells (20 wells for each dilution), 4 negative control wells, and 1 positive control well. (C) Process of elimination used to identify genetically intact, and potentially replication-competent, HIV-1 proviruses. This figure has been modified from Hiener et al.2. Please click here to view a larger version of this figure.
The FLIPS assay amplifies and sequences single, near full-length HIV-1 proviruses. The protocol involves 6 steps to obtain near full-length proviral sequences. These steps include: lysis of infected cells, nested PCR of full-length (intact and defective) HIV-1 proviruses, DNA purification and quantification, sequencing library preparation, NGS, and de novo assembly of sequenced proviruses. The end of each step can be considered a checkpoint in which the quality of the product (e.g. amplified DNA, purified DNA, sequencing library or sequence) can be assessed prior to the next step. An overview of the assessment performed at the end of each step and the expected results is outlined below.
Following nested PCR, amplified products are run on a 1% agarose gel (Figure 3). The initial quality of the PCR can be determined by inspection of negative and positive controls. Negative control wells containing amplified product indicate contamination and positive control wells absent of amplified product indicate insufficient amplification. Next, wells containing amplified product are selected for sequencing. To avoid wells containing mixtures of multiple amplified proviruses, only wells containing amplified product run at end-point dilution are considered for sequencing. According to Poisson distribution, the end-point dilution is found when 30% of wells are positive for amplified product. At this dilution, there is an 80% chance these wells contain a single amplified provirus. Additionally, wells containing multiple amplified proviruses of different lengths can be visualized at this stage as multiple bands will appear on the gel. These wells should not be selected for sequencing (Figure 3).
Following purification of the amplified proviruses selected for sequencing, quantification ensures no proviral DNA is lost during the purification stage. If the DNA concentration of an amplified provirus is <0.2 ng/µL, the remaining sample in the PCR2 plate can be purified. A similar checkpoint occurs following library preparation, in which each individual library is quantified. This ensures individual proviruses have been appropriately fragmented, tagged and amplified prior to sequencing. Individual proviral libraries are pooled in equimolar amounts to a final library concentration of 4 nM (or as specified by the sequencing provider). If the concentration of an individual proviral library is too low, it can be excluded from the sequencing library pool, or the individual library prepared again. A final check of the concentration of the pooled library is performed prior to sequencing along with confirming the average fragment lengths.
Quality control steps before the de novo assembly stage ensures the quality of the reads used to assemble the final proviral contig. These steps include: removal of adapter sequences, trimming of 5' and 3' nucleotides, a stringent quality limit, and disregarding short reads. CLC Genomics Workbench can provide quality control reports that can be used before to assess the initial quality of the reads and guide trimming settings, and then after to determine if the trimming was sufficient to remove low quality regions. Additionally, for de novo assembly, the quality of the assembled contigs can be assessed for sufficient depth (>1000X) and evenness of coverage (Figure 4A). Mixed populations can also be identified at this stage. Mixtures of multiple full-length (~9 kb) proviruses are identified through the presence of multiple SNPs with a frequency of greater than 40%, whereas mixtures of short (containing a large internal deletion) and full-length proviruses can be identified by uneven read coverage following mapping to a full-length reference from the same participant (Figure 4B).
Depending on the application, the final alignment can be visualized using tools such as ggtree available as a package in "R: A language and environment for statistical computing"27. In a recent study, FLIPS was utilized to sequence HIV-1 proviruses from naïve, central, transitional, and effector memory CD4+ T cells isolated from individuals on long-term ART, with the aim to identify if particular cell subsets showed higher proportions of genetically intact and potentially replication-competent HIV-12. Here, visual representation of the sequences isolated from one participant of this study (participant 2026) is presented (Figure 5). In this participant, the majority (97%) of sequences were defective, with intact sequences found in effector and transitional memory CD4+ T cells. This visualization tool is useful for showing the number of sequences with large internal deletions and their position in the genome. It can be annotated further to indicate sequences with deleterious stop codons, frameshift mutations, and deletions/mutations in the MSD site and/or packaging signal.
One application of the FLIPS assay is the identification of genetically intact and potentially replication-competent HIV-1 proviruses. In a recent study of 531 sequences isolated from CD4+ T cells from 6 participants on long-term ART, 26 (5%) genetically intact HIV-1 proviruses were identified2. The remaining defective proviruses included those with inversion sequences (6%), large internal deletions (68%), deleterious stop codons/hypermutation (9%), frameshift mutations (1%), and defects in the packaging signal and/or mutations in the MSD site (11%).
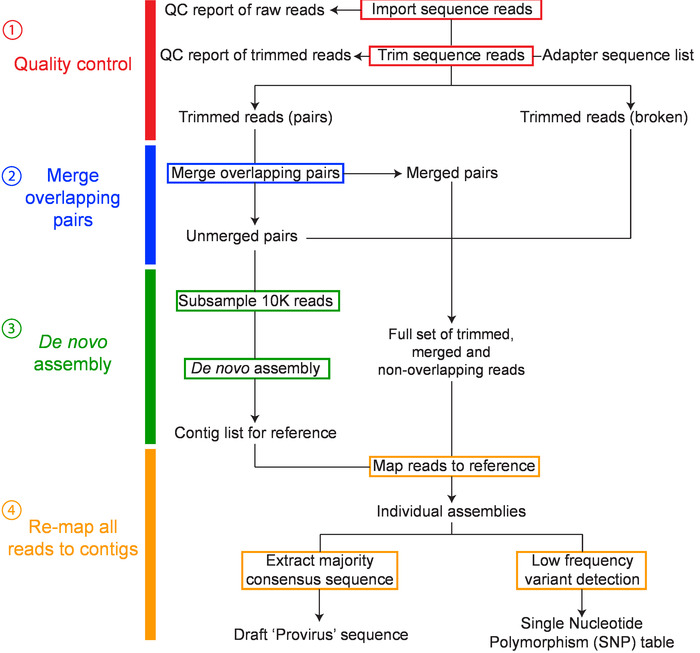
Figure 2: Overview of workflow for de novo assembly of HIV-1 proviruses. The major steps in the workflow include: 1) sequence read quality control, 2) merging overlapping pairs, 3) de novo assembly, and 4) remapping and consensus building have been colored red, blue, green and orange, respectively. This figure has been modified from Hiener et al.2. Please click here to view a larger version of this figure.
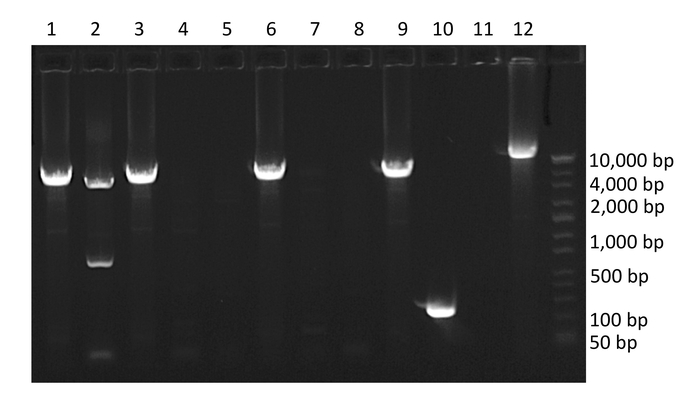
Figure 3: Example agarose gel of PCR amplified HIV-1 proviruses. Wells 1, 2, 3, 6, 9 and 10 contain amplified HIV-1 proviruses. Well 10 contains a provirus with a large internal deletion, well 2 contains co-amplification of two HIV-1 proviruses of different lengths (mixture), and well 12 contains positive control. Note the percent of wells containing amplified product is 60%, which is above the percentage required to isolate single templates. Please click here to view a larger version of this figure.
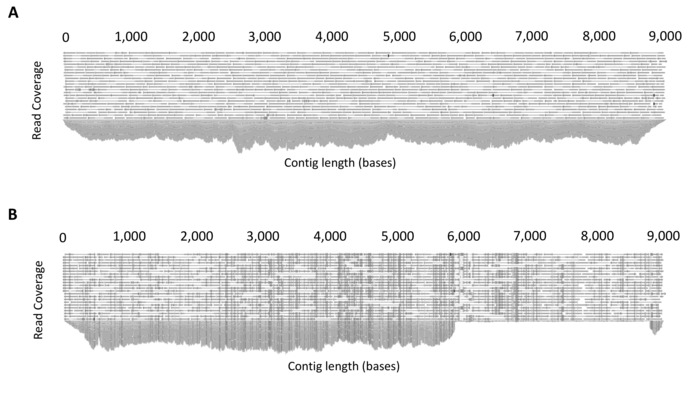
Figure 4: Example output of read mapping. (A) Example demonstrating even coverage due to the amplification of a single full-length HIV-1 provirus. Following de novo assembly, all reads are mapped to the assembled contig to produce a consensus sequence. The software platform allows the mapped reads to be inspected for sufficient and even coverage. (B) Example demonstrating co-amplification of two HIV-1 proviruses of different lengths (mixture). To determine mixtures, reads are mapped to a full-length (~9 kb) reference sequence from the same participant and read mapping inspected. The presence of uneven coverage indicates a mixture. This figure is reproduced with permission from Qiagen14. Please click here to view a larger version of this figure.
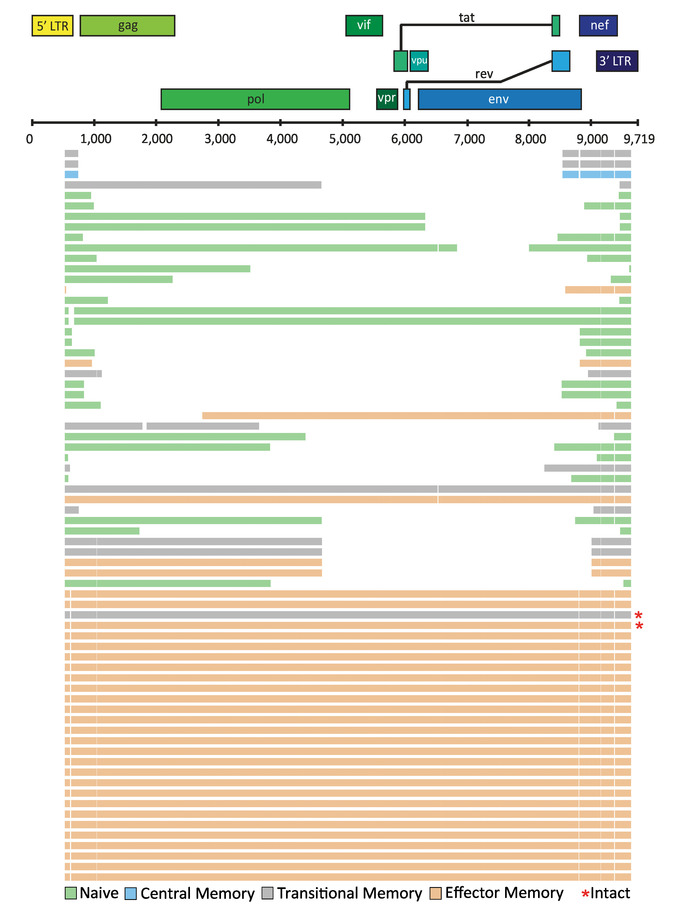
Figure 5: Example visualization of HIV-1 proviral sequences isolated from CD4+ T cell subsets from an individual on long-term ART. Individual HIV-1 proviral sequences are represented by horizontal lines. This figure has been modified from Hiener et al.2. Please click here to view a larger version of this figure.
