Representative results to illustrate the method were obtained for rotenone (Figure 1), paraquat (Figure 2), oxaloacetate (Figure 3), and 4 compounds that were picked up as firefly luciferase inhibitors during screening of a drug library13 (Figure 4). The drug exposure was started at the L4 stage in all cases, but the paraquat exposure experiments were started at 41 hr after food first provided to nematodes compared to 45-46 hr for the other compounds. The protocol allows for a degree of flexibility in the selection of time for the initiation of the drug exposure and the exposure length. However, for screening purposes, set times should be adhered to once selected, for reproducibility between experiments. Endpoints should be measured by 66-67 hr developmental time in order to prevent extensive egg laying and hatching of progeny in wells. Guidelines for timings are provided in Table 1.
Rotenone, a mitochondrial complex I inhibitor, reduced bioluminescence after both relatively short (2 hr) and longer exposures (24 hr) to a range of concentrations (Figure 1). In this instance, no GFP measurements were taken, but the number of technical replicates used (n=6) is sufficient to even out any differences in worm numbers between wells3. The 24 hr exposure is a window of time over which nematodes grow, and slower development as a result of complex I inhibition was expected to contribute to the decline in bioluminescence. This was confirmed by visual observation under stereoscope with delayed development seen, particularly at concentrations of 20 μM and above; in addition some lethality was noticed from 40 μM (qualitative observations not quantified). No lethality was observed after 2 hr exposure but effects on worm movement were seen. A sharp decline in bioluminescence relative to controls occurred at the lowest concentration of rotenone tested 2.5 μM, for which effects were not easily detected by quick visual observation at 2 hr exposure. This decline in bioluminescence is consistent with reduced cellular ATP. Maximal inhibition was achieved with the lowest concentration of rotenone used (2.5 μM) indicating that any subsequent experiments specifically targeting rotenone should be carried out between 0 and 5 μM [the concentration range 0-160 μM was selected as part of a comparison with other drugs initially identified as having significant effects at 10 μM (to be published elsewhere)].
The herbicide paraquat affects mitochondrial function through increase of reactive oxygen species. Here, we demonstrate its effect on bioluminescence of C. elegans strain PE254 after exposure to a range of concentrations for 24 hr (Figure 2). As the strain carries a luc::GFP fusion, GFP fluorescence was also measured as a means of normalization.6,9 Paraquat decreased bioluminescence, GFP fluorescence and normalized bioluminescence significantly. The Dunnet post-hoc test indicated that the concentrations of paraquat with significant differences in relation to vehicle control were 4 and 8 mM for bioluminescence and normalized bioluminescence, as well as 2 mM for normalized bioluminescence. The only concentration significantly reducing GFP fluorescence was 8 mM, a concentration at which growth effects and the occasional dead worm were seen (qualitative observations). The decline in bioluminescence (and normalized bioluminescence) was greater than that of fluorescence, consistent with decreased mitochondrial function and effects on ATP production.
The citric acid cycle intermediate oxaloacetate tested on C. elegans strain PE254 at a single concentration of 8 mM, led to an increase in bioluminescence (Figure 3). No GFP fluorescence measurements were obtained or additional visual observation carried out; however such response to a single concentration would merit further confirmatory exposure to a range of concentrations, and more detailed observations of effects. An enhancement of bioluminescence by this compound is not surprising as it can be postulated to lead to greater activity of the citric acid cycle, with ultimately greater production of ATP (nevertheless it would be advisable to control for any effects on luciferase levels by assessing GFP fluorescence in subsequent experiments). The oxaloacetate data sets shown reveal the extent of variability in response seen in luciferase based experiments. In our experience this variability is a feature of the test system particularly for less detrimental exposure conditions.
The firefly luciferase inhibitory compounds DDD00001434, DDD0001477, DDD00000635 and DDD00023047 were tested both in vitro by looking at effects on the purified enzyme and in vivo using the C. elegans luc:GFP expressing strain. There was no evidence that any of the compounds tested caused nematode death under the exposure conditions. All 4 compounds affected the luciferase activity in vitro at the 2 concentrations tested 25 and 100 μM (Figure 4A). DDD00001434 killed the activity of the purified luciferase almost completely, which provided a credible justification for the large decline in in vivo bioluminescence (Figure 4B). Although, the in vitro and in vivo assays are not directly comparable, the response to DDD00023047 was similar in both assays. DDD0001477 and DDD00000635 caused a greater decline in vivo than in in vitro. These compounds were provided to the live worms 22 hr prior to the luciferin substrate, and results may reflect that they were not easily displaced from the luciferase active site when luciferin became available. Alternatively, DDD0001477 and DDD00000635 may have an additional effect on cellular ATP levels. This would have to be assessed by means that do not involve firefly luciferase. Of note was the strong enhancement of GFP signal in live worms exposed to compound DDD00000635. This will be discussed below.
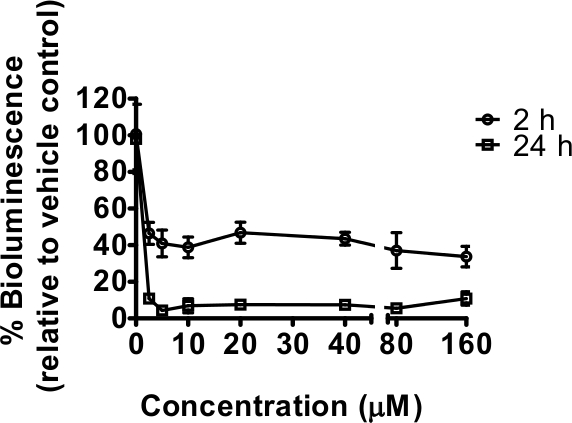
Figure 1. The mitochondrial complex I inhibitor rotenone decreased the energy status of C. elegans as measured by bioluminescence of strain PE254. Synchronized L4 stage worms (45-46 hr after food provided to L1 larvae) exposed to 0, 2.5, 5, 10, 20, 40, 80 and 160 μM rotenone in 1% DMSO for 2 hr (short) or 24 hr (long exposure) prior readings of bioluminescence respectively at 48 and 69 hr of worm development after food provided. Bioluminescence (unit Relative Light Units, RLU) was expressed as a percentage of the vehicle (1% DMSO) values. Error bars depict SEM of technical replicates at each rotenone concentration (n=6). All concentrations tested resulted in a highly statistically significant difference in relation to vehicle control (p<0.001).
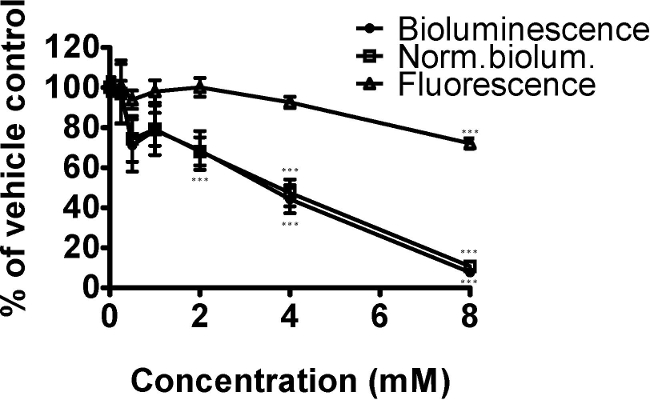
Figure 2. The oxidative stressor paraquat decreased the energy status of C. elegans strain PE254 in a concentration dependent manner (measured by bioluminescence). Synchronized L4 stage worms (for convenience in this instance at 41 hr after food provided to L1 larvae) were exposed to 0, 0.25, 0.5, 1, 2, 4 or 8 mM paraquat for 24 hr. GFP fluorescence (unit Relative Fluorescence Units, RFU) was used to normalize bioluminescence data (unit RLU), both endpoints obtained at 65 hr development. Data are expressed as a percentage of controls (1% DMSO). Error bars depict SEM of technical replicates (n=5 for different concentrations; n=18 for controls). One-way ANOVA: *** p<0.001 relative to vehicle control.
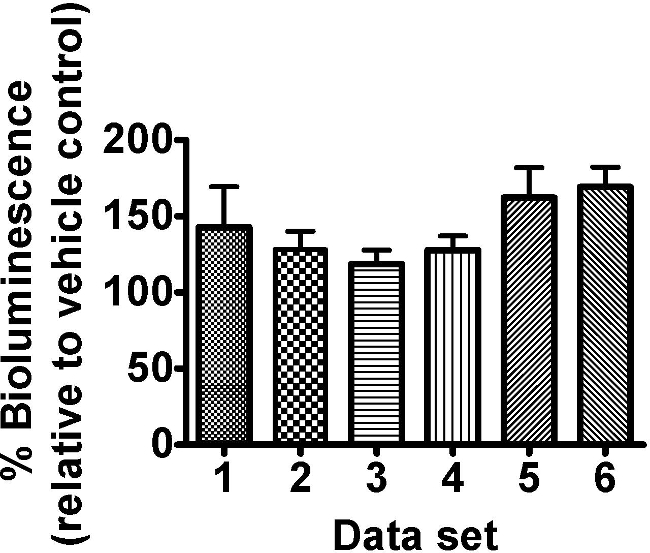
Figure 3. The citric acid cycle intermediate oxaloacetate (8 mM) enhanced the bioluminescence of C. elegans strain PE254. Synchronized L4 stage worms (45-46 hr after food provided to L1 larvae) were exposed to freshly prepared 8 mM oxaloacetate for 18 hr ± 30 min. Bioluminescence (unit RLU) was read at a developmental time of 63.5 hr ± 1 hr and was expressed as a percentage of the vehicle control (ddH2O). Different columns represent different data sets of 8 technical replicates allocated to different 96 well plates within the same experiment. Error bars depict SEM of technical replicates (n=8). 2 way-ANOVA with ‘set’ and ‘concentration’ as factors: ‘Set’ p>0.05, ‘concentration’ p<0.001 and interaction term p>0.05.
A
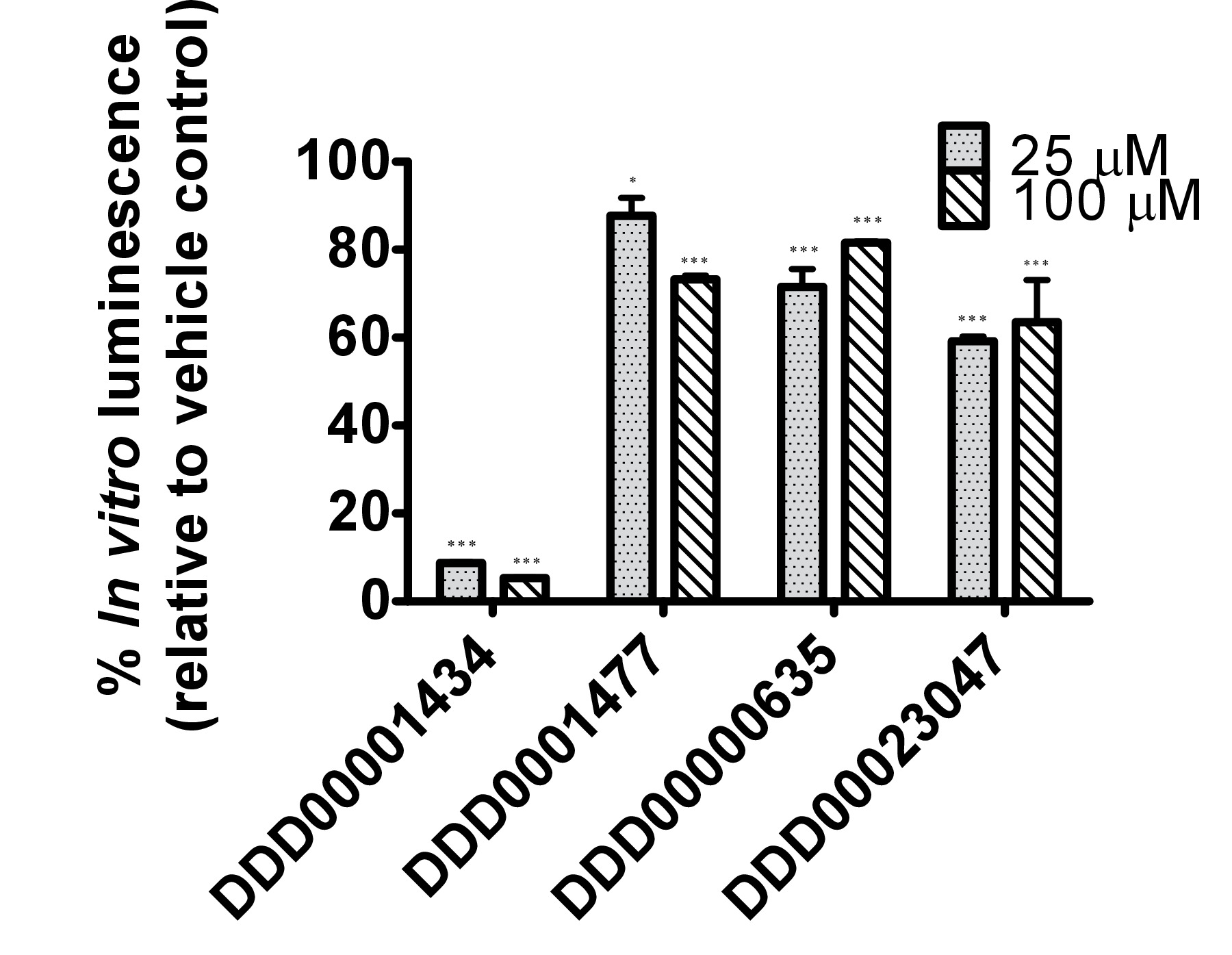
B
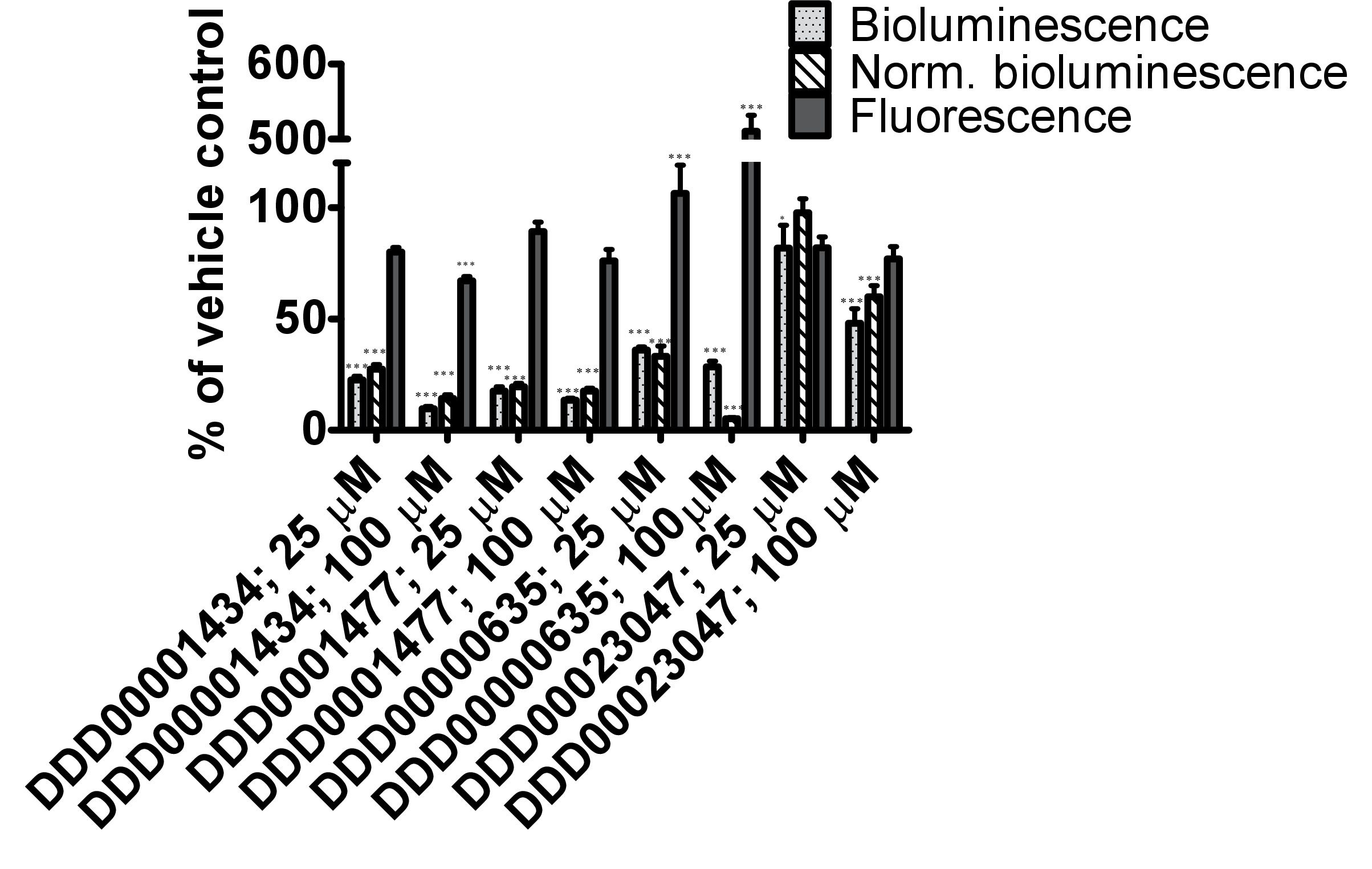
Figure 4. Testing of responses to 4 compounds from the Dundee drug discovery compound library13 (C1:DDD00001434, C2:DDD0001477, C3:DDD00000635 and C4:DDD00023047) with known inhibitory activity on firefly luciferase luminescence. (A) Effect of compounds on the activity of purified firefly luciferase (measured as light units, RLU), expressed as a percentage of vehicle control. The ATP bioluminescence CLSII kit (Roche, Manheim, Germany) was used according to instructions with the same amount of luciferase enzyme aliquoted to wells (white 96-well microtiter plates). ATP was provided at a final concentration of 10 μM and 1 μl of compound (final concentration indicated) or DMSO (1%) was added. Luminescence signal was integrated for 10 sec. Error bars depict SEM of technical replicates (n=4). Analysis: One-way ANOVA; *p<0.05 or *** p<0.001 relative to vehicle control. Differences between 25 and 100 μM highly significant for C1 (DDD00001434; p<0.001), and significant for C2 and C3 (respectively DDD0001477 and DDD00000635; p<0.05). (B) In vivo measurements of bioluminescence, bioluminescence normalized to GFP fluorescence and GFP fluorescence of C. elegans strain PE254 at 67 hr developmental time following exposure to test compounds for 22 hr. Error bars depict SEM of technical replicates (n=8). Statistical analysis (One way-ANOVA) carried out on pooled data from 3 data sets (3 x n=8). *p<0.05 or *** p<0.001 relative to respective vehicle control. Differences between 25 and 100 μM only statistically significant (p<0.05) for normalized bioluminescence following exposure to C4 (DDD00023047).
| Time of day | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | Day 8 |
| 9-10 | Step 5 | |||||||
| 10-11 | Step 5 | |||||||
| 11-12 | Step 4 | Step 5 | ||||||
| 12-13 | Step 4 | |||||||
| 13-14 | ||||||||
| 14-15 | ||||||||
| 15-16 | ||||||||
| 16-17 | Step 1 | Step 3 | ||||||
| 17-18 | ||||||||
| 18-19 | Step 2 |
Table 1. Overview of protocol steps to be carried out on each day of nematode experiments (Section 3) and suggested timings.
