
Figure 1: Standardization of Neuronal Cultures using Microfluidic Devices7. (a) Device assembly: when the microfluidic devices are properly assembled on a dry surface all chambers are visible. (b) Cell plating: plate the cells on the top right well and cells should move towards the left well. (c) Cell density: just after plating, check in the microscope if the concentration of cells is adequate. (d) After 1 d in culture, hippocampal neurons are well adhered close to the microchannels and start forming neurites.
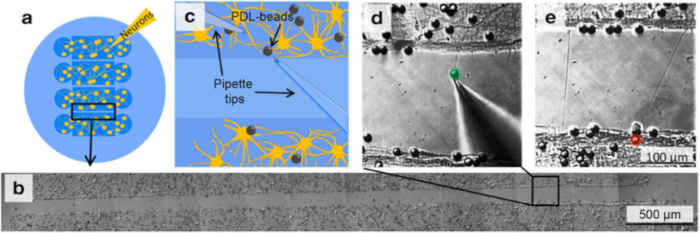
Figure 2. Initiation, Elongation and Connection of New Neurites to Connect two Isolated Populations using Multiple Population Device and Micromanipulation. (a) The device isolates 4 neuronal populations between 3 gaps of 100 or 200 µm each. (b) Following removal of the device, select one gap and certify that no neurites connect the 2 individual populations. (c) Schematic of the experimental set-up as should be visible in the optical microscope, indicates the position of two micropipettes and the presence of PDL-coated beads. (d) By applying negative pressure to a pipette, a PDL-bead adhered to one neuronal population is pulled with the pipette tip, thereby initiating a new neurite. By maintaining the negative pressure in the pipette, the PDL-bead-neurite complex (green) can be pulled, elongating the neurite. (e) Pipette micromanipulation guides extension of the new neurite over the gap and the formation of a connection with a new neuronal population. To ensure the adhesion of the new neurite to the second population, a PDL-bead (red) is positioned with a second pipette on top of both the extended neurite and neuronal population.

Figure 3: Maintenance of healthy neuronal cultures for several weeks. (a) Add medium every 2-3 d and keep a positive meniscus in the upper wells of the microfluidic chambers so cells will have a constant supply of nutrients. (b) Keep cells inside a larger plate with a dish containing water to reduce medium evaporation. (c) Use sterile tip and tweezers to (d) easily peel off the microdevices.

Figure 4: Location of Pipette Depositing Beads into Cultures. Once the microfluidic device has been removed, neurons are visible on the coverslip. When depositing beads, position the pipette tip so that it is in the center of the cells.
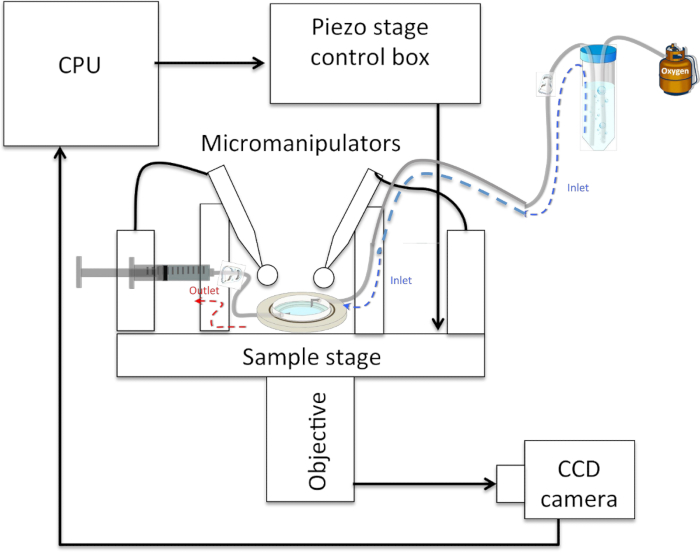
Figure 5: Schematic of a Typical Neurite-pulling Set-up. The sample rests on a (piezo-actuated) stage and can be accessed from above by 2 micropipettes held in micromanipulators and connected to 1 mL syringes via plastic tubing. The sample is accessed optically from below by an objective connected to a CCD camera that sends images to a CPU. An inlet tube feeds oxygenated physiological saline solution to the sample, which rests below it, and an outlet tube connected to a syringe allows the withdrawal of solution in the event of overflow.
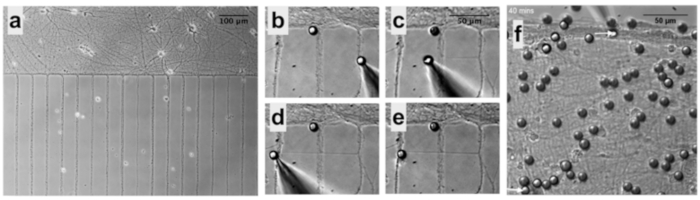
Figure 6: PDL-bead Adhesion to Neurons and Pipette Micromanipulation. (a) Neurons remain organized in patterns after removal of microfluidic chambers allowing easy identification of soma and neurites. (b) Micromanipulation of PDL-beads adhered to neurites enables neurite initiation, extension (c) and connection (d), followed by release of the PDL-bead from the pipette (e). (f) After contacting two isolated neuronal populations (bottom arrow), a second PDL-bead and pipette micromanipulator (top arrow) is used to establish a second adhesion point, a few hundreds of microns apart form the first contact point, and guarantee functional connections.
