Source: Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland School of Medicine and the Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
ELISPOT is a standardized, reproducible assay used to detect cellular immune responses. The assay utilizes an enzyme-linked immunosorbent assay (ELISA)- based method to detect single-cell immune responses which can be visualized by spots, hence the name ELISPOT. ELISPOT was first described in 1983, by Czerkinsky, as a method of enumerating the number of B cell hybridomas producing antigen-specific immunoglobulins (1). The same group further developed the assay to measure the frequency of cytokine producing T lymphocytes. Now ELISPOT has become a gold standard for measuring antigen-specific T cell immunity in clinical trials and vaccine candidates. For example, after vaccination or during an infection, plasma cells and memory B cells secrete antibodies that provide protection. Typically, these B cell responses are assessed by measuring serum titers of antigen-specific antibodies. However, this type of analysis, typically measured by ELISA, may not include memory B cells, which can be present even in the absence of detectable serum antibody levels. Furthermore, it has been well-established that circulating memory B cells are important for the rapid and protective antibody response observed following pathogen re-exposure, thus it is critical to be able to detect these cells. Therefore, to clearly assess antigen-specific memory B-cell responses, both ELISA and ELISPOT should be used (2).
ELISPOT assay uses a plate containing membrane-lined wells that are coated with antibodies in order to capture secreted proteins of interest. Then, the plate is loaded with cells and stimuli to induce protein production. The secreted proteins are captured by the antibodies coated on the surface. After appropriate incubation time, cells are removed and the secreted molecule is detected by using a biotinylated antibody that is specific for a different epitope, as compared to the capture antibody. Next, streptavidin peroxidase is added, followed by the addition of a substrate that allows the detection of the spots (Figure 1). The strength of this assay is that it allows one to quantitate the number of cells producing the protein of interest. Importantly, one can assess if there are changes in the total number of cells producing a specific protein or if individual cells within a population are producing more protein. Moreover, it can provide information regarding kinetics and can be used to assess overall immune activation (mitogen stimulation) relative to antigen-specific responses (antigen simulation). The ELISPOT assay will permit the detection one activated cell amongst 300,000 cells following mitogenic or antigen-specific activation.
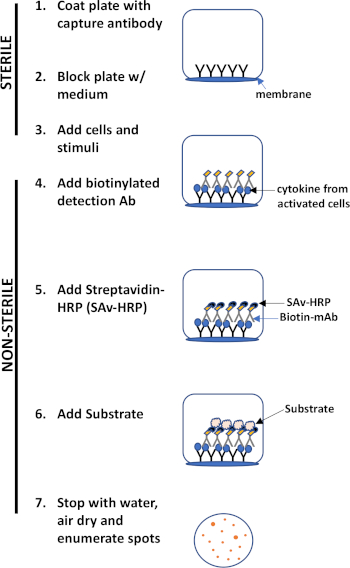
Figure 1: ELISPOT protocol overview.
The major advantages of this assay are its- a. Simplicity- the protocol is relatively simple and straightforward. It does not require technical expertise, b. Sensitivity- it permits the detection of immune cells at the single cell level and requires very few cells compared to other methods such as flow cytometry, c. Functionality- it provides quantitative data regarding immune function.
This lab exercise demonstrates the ELISPOT protocol for detection of IFN-γ secreting-splenocytes, but as mentioned above this assay can also be used to assess antibody secretion by B cells (3).
In this ELISPOT assay, splenic leukocytes from wildtype and tumor-bearing mice were analyzed for IFN-γ. Figure 2 A shows the visual image of the assay result. The numbers in the green color indicate the number of spots per well (TNTC indicates “too numerous to count”). Notice that the number of spots decreases with decreasing cell concentration.

Figure 2A: Decreased immune responses in tumor-bearing mice. Please click here to view a larger version of this figure.
Typically, ELISPOT data are presented as the number of spot counts per number of cells plated. In Figure 2 B the number of spots is displayed in a bar graph, with each respective cellular concentration listed on the X-axis. For graphing purposes, 150 was used to indicate the maximum number of spots. The number of IFN-γ producing murine splenic leukocytes in tumor-bearing animals is lower than the wild type ones.

Figure 2B: Decreased immune responses in tumor-bearing mice. Splenocytes were harvested from control C57BL/6 (wildtype) and tumor-bearing mice and stimulated with PMA/ionomycin for 48 hours. ELISPOT assays were used to quantitate the number of IFN-γ-producing splenic leukocytes. (A) Visual and (B) graphical representation of the data. TNTC indicates too numerous to count. For graphing purposes, 150 was used to indicate the maximum number of spots. The green numbers indicate the number of spots counted per well. The red numbers indicate the reference wells that were used to determine which spots were cells and which spots were debris, artifacts, or edge effects and should be excluded from the analysis.