1. NK-EV biomanufacturing from NK92-MI cells using a closed-loop bioreactor
NOTE: NK-EVs are manufactured using a scalable biomanufacturing workflow that adheres to Good Manufacturing Practices (GMP) and utilizes the NK92-MI cells (see Figure 1). Our recent publication has detailed insights into the biomanufacturing procedure and NK-EV products' identity and safety profiles7.

Figure 1: Biomanufacturing of natural killer cell-derived extracellular vesicles (NK-EVs) in a closed-loop hollow-fiber bioreactor (HFB) with scalable isolation workflow. Schematic representation of the biomanufacturing workflow to generate large quantities of high-purity NK-EV products. IL-2 self-sufficient NK92-MI cells are seeded into a closed-loop HFB cartridge and cultured under serum-free (SF), xeno-free (XF), feeder-free, and antibiotic-free conditions, where they are grown for continual EV-rich conditioned medium collection. NK-EV isolation from EV-rich CM is performed by Fast Protein Liquid Chromatography-based size exclusion chromatography (FPLC-SEC) coupled with ultrafiltration (UF). NK-EVs are characterized and assessed through multiple assays, and their functionality against K562 leukemia cells is evaluated using a viability potency assay. This figure has been modified from7(created with Biorender.com). Please click here to view a larger version of this figure.
- Starting from 1 – 5 x 106 NK92-MI cells and maintaining a cell density between 3 – 8 x 105 cells/mL, culture the cells in T25 – T175 flasks using a pre-warmed culture medium. Incubate at 37 °C with 5% CO2 (see Table of Materials). Replace the medium every 2 – 3 days until 1 x 108 NK92-MI cells are produced of at least 70% viability.
NOTE: Keep 1/5th-1/3rd of the conditioned medium (CM) when reseeding the cells, as it contains favorable growth factors. - Perform medium HFB cartridge preparation and NK cell inoculation as described below.
NOTE: All manipulations should be performed inside a class II biosafety cabinet to ensure and maintain sterility. Before moving the cartridge system into the biosafety cabinet, generously spray with 70% ethanol, paying special attention to the reservoir bottleneck and the syringe connections.- Prepare the HFB cartridge according to the manufacturer's instructions19 (see Table of Materials; see Figure 2) as described below.
- Wrap Luer Lock connections with wax film and adjust the pump flow rate according to the manufacturer's instructions19. Condition the HFB cartridge by allowing 150 mL of sterile phosphate-buffered saline (PBS; see Table of Materials) to circulate for at least five days.
- To remove the air from the extracellular capillary space (ECS; volume is approximately 29 mL), inject approximately 40 mL of PBS through the left ECS port and allow the air to escape through the right ECS port. While doing that, close the left and right end port clamps. Ensure the syringe is always connected to the left and right ECS ports.
- Once completed, put the cartridge on the flow pump (see Table of Materials) inside the incubator set to 37 °C and 5% CO2. Ensure there are no leaks after a few days of circulation.
- Replace the PBS with 150 mL of culture medium in the reservoir bottle and the ECS 2 days before seeding cells into the cartridge. Repeat the previous conditioning steps (step 1.2.1.) using the culture medium but for 2 days of circulation.
- Before seeding cells, replace the contents in the reservoir bottle and the ECS with 250 mL of fresh culture medium.
- Acquire the culture flask from the incubator and transfer the cells to a 50 mL tube. Spin at 300 x g for 5 min. Resuspend the cell pellet using 21 mL of culture medium.
- Prepare two aliquots of 20.5 µL each from the cell suspension for cell counting on an automated cell counter (see Table of Materials). To each 20.5 µL cell suspension aliquot, add an equal amount of AO/PI dye (see Table of Materials) and mix up and down at least 10x.
NOTE: We do not recommend Trypan Blue for accurate NK cell counting. Alternatively, use a hemocytometer for manual counting. - Load 20 µL into each counting chamber of the counting slide and perform automated cell counting using the appropriate program. Calculate the average live cell concentration and note viability.
- Mix the NK cell solution a few times before aspirating it using a 20 mL syringe and an 18 G needle to maintain sterility. This solution should contain approximately 1 x 108 live NK cells in approximately 20 mL or about 5 x 106 cells/mL.
- After removing the needle from the syringe, gently inject the NK cells into the cartridge through the left ECS port. To ensure uniform cell dispersion throughout the cartridge, gently reciprocate the cell solution at least 10x using the syringes connected to the left and right ECS ports.
NOTE: the solution should have equal turbidity across both syringes, with the left and right end ports closed. - Open the left and right end port and inject what remains within the syringes. Close the left and right ECS ports using the clamps.
- Transfer the cartridge to the incubator and let it sit for 30 min before properly installing it on the flow pump. Leave the cartridge in for biomanufacturing. Adjust the flow rate according to the manufacturer.
- To monitor cell health metrics, acquire a 0.5 mL aliquot of medium daily from the thoroughly mixed medium in the reservoir bottle and store it at -20 °C after verifying glucose and pH levels. L-lactate levels can be verified later (see Table of Materials).
- Replace the medium in the reservoir (250 – 500 mL) every 1 – 2 days to maintain the glucose content above 50% of the initial levels found in the medium and pH above 7.0 (range from 7.0 – 8.0).
- Prepare the HFB cartridge according to the manufacturer's instructions19 (see Table of Materials; see Figure 2) as described below.
- Perform NK-EV-rich CM collection daily after 1 day of rest when first seeding the cartridge as described below.
- Move the cartridge system into the biosafety cabinet. Gently inject approximately 21 mL of culture medium through the left ECS port to push an equivalent volume of EV-rich CM through the right ECS – do not mix (see Figure 3).
NOTE: Always use new plasticware to prevent contamination. - Transfer EV-rich CM solution into a 50 mL tube and centrifuge at 300 x g for 5 min. Meanwhile, move the cartridge system back into the incubator on the flow pump.
- Transfer the supernatant to a new tube and centrifuge at 2000 x g for 10 min. Again, transfer the supernatant into a new tube. Then, aliquot the EV-rich CM equally across 3, 50 mL tubes (~7 mL/tube) and store at −80 °C until further processing.
NOTE: Sequentially harvested EV-rich CM is pooled across these three tubes, generating three technical replicate sample tubes.
- Move the cartridge system into the biosafety cabinet. Gently inject approximately 21 mL of culture medium through the left ECS port to push an equivalent volume of EV-rich CM through the right ECS – do not mix (see Figure 3).
- Perform HFB-NK cell harvest to continue producing EV-rich CM using the same HFB cartridge as described below.
NOTE: NK cells can be harvested from the HFB's ECS by performing the HFB-NK cell harvest protocol" once the cartridge reaches confluence (maximum of 1 x 109 cells). This happens after 5 – 7 days for each lot or when the glucose content is found to be below the limit of detection of the glucose meter (e.g., no reading or readings of ~ 0) for 2 consecutive days. If this is the final cell harvest, the medium can be substituted for PBS to flush the cartridge and retrieve the cells.- Harvest EV-rich CM exactly as detailed above in step 1.3.
- Inject approximately 50 mL of the medium through the left ECS port. To ensure homogenous cell dispersion throughout the cartridge, gently push back and forth the cell solution using the syringes connected to the left and right ECS ports at least 10x to loosen up cells before ultimately pushing and collecting them with a syringe through the right ECS port. Transfer the harvested EV-rich CM into a 50 mL tube. Set aside at 37 °C (water bath or incubator) for now.
NOTE: The push-back action helps to dislodge the cells before they are fully expelled and collected by a syringe through the right ECS port. The solution should have equal turbidity across both syringes, with the left and right end ports closed. Tapping on the bioreactor cartridge (physical disturbance) can help preemptively dislodge the cell cluster at the bottom of the cartridge. Aggressive back-forth mixing of the cell suspension can negatively affect the viability of the recovered cells. Care and patience should be applied to maximize viability. - Repeat the last step 2x. In total, 150 mL of cell suspension should be recovered. Centrifuge at 300 x g for 5 min. Discard supernatant.
- Resuspend both cell pellets in 20 mL of fresh medium each and combine them. Collect two aliquots of 20.5 µL each of the cell suspension for cell counting on an automated cell counter (see Table of Materials).
NOTE: Typically, numerous cell dilutions using PBS as diluent are required to fall within the cell counter's dynamic range. - To the 20.5 µL cell suspension aliquot, add an equal amount of AO/PI dye (see Table of Materials) and mix up and down at least 10x. Load 20 µL into each counting chamber of the counting slide and perform automated cell counting using the appropriate program.
- Average the live cell concentration of all dilution-corrected counts, determine the total amount of live cells, and record the viability. As detailed above, to continuously produce EV-rich CM using the same bioreactor cartridge, reseed 1 x 108 HFB-produced NK cells.
NOTE: If desired, HFB-produced NK cells can be stored using a cryopreservation freezing medium and a freezing container to control the freezing rate (see Table of Materials).

Figure 2: Hollow-Fiber Bioreactor (HFB) system component and set-up. The reservoir bottle (1) contains the complete medium that circulates through the bioreactor cartridge (2) by the action of a peristaltic pump (not shown) acting on the pump tubing (3). Cells are introduced into the extracellular capillary space (ECS) through the left (4) and right (5) ECS side ports. Once the ECS slide clamps are closed, the left (6) and right (7) end side ports are open to allow the medium to circulate throughout the system. Notice the addition of wax film on the Luer Lock connection near the reservoir cap of the medium bottle to prevent potential contamination. Please click here to view a larger version of this figure.

Figure 3: Schematic representation of the NK-EV isolation process. After daily collection of EV-rich conditioned medium (CM), the solution was differentially centrifugated to remove cells (first spin at 300 x g for 5 min) and cellular debris (second spin at 2000 x g for 10 min). Cleared EV-rich CM was stored at -80 °C until further processing. Once ready for NK-EV isolation, frozen EV-rich CM is thawed and centrifuged one more time to ensure the removal of cellular debris (third spin at 10,000 x g for 30 min). Then, the EV-rich CM is treated for 2 – 4 h at 37 °C with endonuclease to digest nucleic acids considered host cell contaminants. Next, the EV-rich CM is processed by Fast Protein Liquid Chromatography-based size-exclusion chromatography (FPLC-SEC) for EV purification using a bimodal resin. Eluted fractions of approximately 10 – 15 mL are combined and filtered with 0.22 µM filters to ensure the sterility of the final NK-EV product. Ultrafiltration allows the product to be concentrated by a factor of about 35 – 50X, yielding a guaranteed concentration of over 1 x 1012 EVs/mL, totaling 1.0 – 1.5 mL. This figure has been modified from7(created with Biorender.com). Please click here to view a larger version of this figure.
2. NK-EV purification by FPLC-SEC coupled with UF and filter-based sterilization
- Prepare the following solutions and filter them twice using a 0.1 µm filter (see Table of Materials): water (conductivity of 0 mS/cm), PBS: 50 mL of 10x PBS + 450 mL of water (conductivity around 14.7 mS/cm), 20% ethanol, cleaning solution (0.5 M NaOH and 30% isopropyl alcohol in water).
- Perform FPLC system initiation according to manufacturer's instructions (see Table of Materials). Perform pre- and post-run clean-in-place (CIP) steps according to manufacturer's instructions. Wash all lines and the column resin and rinse using double-filtered (DF) water, cleaning solution, DF-water, and DF-PBS.
NOTE: It is worth noting that CIP can be done on another day if needed. - Use a chromatography column packed with a bimodal resin (see Table of Materials) with a bed height of 20 cm. Make connections using the drip-to-drip method to ensure no air is introduced inside the column.
- Set up the fraction collector with appropriate collection tubes and change the fractionation settings to the desired collection volume (e.g., 15 mL). Place enough tubes and two additional tubes to collect the entire sample volume.
- Carry out sample preparation as described below.
- Take 40 – 80 mL of EV-rich CM from the -80 °C freezer and thaw quickly at 37 °C. Load the sample into the ultracentrifuge and spin at 10,000 x g for 30 min at 4 °C.
NOTE: Tubes must be balanced accurately by weight, not by volume. - After spinning, collect the supernatant and transfer it to a new tube. To reduce dsDNA levels, treat the EV-rich CM with 50 U/mL of endonuclease and 1.5 mM of MgCl2 (see Table of Materials). Incubate for 2 – 4 h in an incubator (37 °C), allowing moderate mixing.
- Take 40 – 80 mL of EV-rich CM from the -80 °C freezer and thaw quickly at 37 °C. Load the sample into the ultracentrifuge and spin at 10,000 x g for 30 min at 4 °C.
- Once the system (lines and column) is ready for EV isolation, load the EV-rich CM into a 60 mL syringe and connect it to the sample line. Start the system by clicking Manual Run and set the flow rate to 0. Follow the software prompts to save the run pre-emptively, then click Start.
- Select Line B (DF-PBS) and run at a flow velocity of 150 cm/h (flow rate of 2.0 mL/min). Make sure the solution runs through the column.
- Once the conductivity stabilizes, press Auto Zero UV. Change the flow path to direct the sample to the waste bottle before the column. Make sure no bubbles are introduced into the system.
- After a maximum of 5 – 20 s, direct the sample to the column. Click Fractionation once the UV readings reach approximately 230 mAU.
- Once the sample is completely injected through the system, switch the buffer system over to DF-PBS (at the sample valve) to continue purification. Click Fractionation again when the UV value reaches approximately 1600 mAU.
NOTE: This corresponds to the intersection between UV readings and conductivity readings. Longer fractionation only dilutes the retentate without increasing EV yield. - Combine all the fractions (diluted NK-EVs) and store them at 4 °C until ready for filter-based sterilization and ultrafiltration (UF).
- Continue running DF-PBS until the UV value reaches approximately 1000 mAU. After this, stop running and save the chromatogram as a PDF document.
3. NK-EV product filter-based sterilization and concentration by UF
- Cool down the centrifuge to 10 °C. Sanitize every component of the UF apparatus (see Table of Materials) by rinsing with 20 – 30 mL of 90% ethanol. Spin at 4000 x g for 5 – 10 min.
NOTE: The filter is made of 10 kDa MWCO regenerated cellulose. - Discard the flow through and then repeat the rinse using sterile PBS to equilibrate the device. Repeat 2x in total.
- To maximize sterility, filter the diluted NK-EV solution using a 0.22 µm syringe filter pre-wet with DF-PBS (see Table of Materials). Collect the filtrate directly into the sterilized concentration apparatus.
- Spin at 4000 x g for 15 – 40 min (spin time is sample dependent). Mix the solution within the top filter compartment using a serological pipette after spinning. Spin at 4000 x g for an additional 10 min.
NOTE: The mixing step is optional as it simply eases the concentration step by preventing the membrane from being clogged by EVs. - Temporarily set aside the flow-through and collect the NK-EV sample by inverting the filtration device and attaching it to the collecting device.
- Spin at 2000 x g for 2 min. Transfer the purified NK-EV product into a 2 mL tube. Store the purified NK-EV product at 4 °C for short-term use (≤ 7 days) or frozen at -20 °C for long-term use.
4. NK-EV characterization by Nanoparticle Tracking Analysis (NTA)
- Prepare the solution and filter it twice at 0.1 µm (see Table of Materials): water, PBS, cleaning solution (10% bleach (CAUTION) in water).
- Initiate the NTA system according to the manufacturer's instructions. Similarly, perform the pre- and post-run clean-in-place (CIP) steps. Wash all lines and rinse using double-filtered (DF) water, cleaning solution, and DF water. Equilibrate the lines using DF-PBS.
- Verify the flow cell and check for air bubbles. Remove bubbles if present. Once clear, carefully re-insert the flow cell into the NTA instrument.
NOTE: Although not recommended by the manufacturer, very difficult-to-remove air bubbles can easily be removed by rinsing with 20% ethanol and then DF-water. - Once the flow cell is in place and the door is closed, click Start Camera. With the lines filled with DF-PBS, the screen should show an absolute minimum number of particles.
- Change capture settings to a screen gain of 2 and a camera level of 14. Also, turn on the heater to temperature-stabilize the flow cell.
- Click Standard measurement to create a script under the SOP tab for collecting one capture over 1 min at a flow rate of 30 particles/frame and 23 °C.
- Just below, add the folder and file name to the pathway name to save the data.
- Prepare dilutions of the purified NK-EV product using DF-PBS in advance. When running NTA, accurate quantification requires 30 – 80 particles/frame.
- Vortex the sample before loading it into the syringe (see Table of Materials).
- Carefully connect the 1 mL acquisition syringe to the instrument loading line. No air should be present as it will negatively affect the acquisition and analysis. Slowly push half of the sample through, leaving around 0.5 mL in the syringe.
- Once particles are visible on the screen, focus the camera to have a maximum of one halo around each particle. Click Infuse under the Hardware tab at a rate of 1000 for 5 s. Then, bring it down to a rate of 30.
- Press Run Script and follow the prompts. The software will ensure the temperature is set and ask if the settings are correct. Click Yes and follow the software prompts.
- After completing the capture, click İptal when the software asks to process or export files. Click Infuse under the Hardware tab at a rate of 1000 for 10 – 15 s. In the meantime, turn back on the heater and the camera. Then, bring the rate down to 30 until the particles move.
- Gather four more captures by repeating the previous steps. Once a total of five captures have been recorded per dilutions, perform analysis after importing all five captures.
- Select the files to be processed by highlighting them. Click Process Selected Files. Under the Process tab, adjust the analysis settings to a screen gain of 2 and a detection threshold of 15.
Settings are sample-dependent; ensure that 30 – 80 particles per frame are visible. - Check and click OK for analysis.
- Once the files are processed, the software will ask to export them. Click Yes without clicking additional boxes or click Export.
- Repeat for all EV dilutions or samples. Shut down the NTA instrument after all samples are completed and CIP is done.
5. Quality assurance testing
- Perform microbial testing using two tests: 1) a small aliquot of purified NK-EVs is spiked into autoclaved LB broth medium, and 2) a small aliquot of purified NK-EVs is used for mycoplasma PCR detection (see Table of Materials).
- Test 1: culture LB medium at 37 °C for up to 5 days of with positive and negative controls included. Record the OD600, if needed.
- Test 2: perform mycoplasma PCR detection according to the manufacturer's protocol.
- Quantify protein and dsDNA on purified NK-EV dilutions using fluorometer-based assays as per manufacturer's instructions (see Table of Materials).
6. Potency evaluation of NK-EV treated cancer cells using a validated highly sensitive resazurin-based cell viability assay 20
- Culture human K562 leukemia cells using RPMI-1640 with 10% heat-inactivated FBS for a few days before performing the potency assay (see Table of Materials). Maintain density between 2 – 8 x 105 cells/mL and replace media every 2 – 3 days.
- Acquire a 96-well flat-bottom plate (see Table of Materials) and pre-emptively add the volume of assay medium (supplemented with 5% EV-depleted FBS) required for normalization purposes (see Table of Materials). The final volume is 150 µL/well.
NOTE: Use a repeater pipettor to reduce well-to-well variation. - Acquire the cell culture and transfer the cells to a tube. Spin at 300 x g for 5 min. Resuspend the cell pellet into a single-cell solution using 2 – 5 mL of assay medium.
- Collect an aliquot of 20.5 µL of the cell suspension for cell counting on an automated cell counter (see Table of Materials).
- To the 20.5 µL cell suspension aliquot, add an equal amount of AO/PI dye (see Table of Materials) and mix up and down at least 10x.
NOTE: We recommend AO/PI for accurate cell counting. Alternatively, use a hemocytometer for manual counting. - Load 20 µL into each counting chamber of the counting slide and perform automated cell counting using the appropriate program. Average the live cell concentration and record the viability.
- Transfer approximately 1 x 106 cells into a secondary tube. Dilute the cells to precisely 7 mL of assay medium and repeat cell counting. The concentration should be approximately 1.2 – 1.5 x 105live cells/mL.
- As detailed above, adjust the single-cell suspension concentration to 1 x 105 live cells/mL and repeat cell counting if needed.
NOTE: The coefficient of variation between technical duplicate counts should be less than 25%; typically, it is less than 5% with AO/PI counting. - Once the desired concentration is achieved, transfer 50 µL (± 1 µL) of this solution into each well to get as close as possible to 5000 cells/well (4900 – 5100 cells/well). Prepare technical triplicates for each assay condition and use a repeater pipettor to reduce well-to-well variation.
- Transfer the plate to an orbital shaker (350 – 500 RPM) for 2 min. Transfer cells back to the incubator until ready to proceed with NK-EV treatment.
- Prepare the required NK-EV dilutions (1:5, 1:10, and 1:100) using an assay medium.
- From these dilutions, test the following EV concentrations: 1 x 108, 5 x 108, 1 x 109, 5 x 109, 1 x 1010, 5 x 1010 and 1 x 1011 particles/mL. The dosing volume is limited to 20% of the total assay volume.
- Once ready, transfer the required volume of a given dilution to the wells requiring a desired EV concentration for treatment. Add 15 µL of 10x Triton-X to the positive control well (see Table of Materials). The final well volume should be normalized to 150 µL.
- Add the plate to an orbital shaker (350 – 500 RPM) for 2 min. Incubate the cells at 37 °C in the 5% CO2 incubator for 3 h.
- Pre-warm the plate reader (see Table of Materials) at 37 °C and load the following script: 37 °C (reduces temperature-related variation), 450 RPM mixing for 1 min (ensures sample homogeneity), and read.
- Add 15 µL of the resazurin-based reagent to each well (see Table of Materials). Protect the reagent from light and use a repeater pipettor to reduce well-to-well variation.
- Add the plate to an orbital shaker (350 – 500 RPM) for 2 min. Transfer the plate to the incubator and incubate for 60 min. Remove air bubbles using an ethanol-dipped pipette tip. Read plate using an excitation of 560 nm and an emission of 590 nm.
- Data analysis: Average technical replicates were averaged and correct for background before performing a dose-response analysis using a non-linear regression for the inhibition effect showing the log(inhibitor) vs. normalized response-variable slope without constraint. Record the Hillslope and EC50 values.
NK-EVs possess inherent cytotoxic functions and have demonstrated high efficacy against various cancer models. However, there needs to be more standardization among current studies regarding a biomanufacturing workflow suitable for the large-scale production of NK-EVs6,21. Our previous study described the feasibility of a closed-looped hollow-fiber bioreactor (HFB) system to produce large quantities of high-purity NK-EV products7. As a follow-up, this protocol-based study details the biomanufacturing workflow and demonstrates its reproducibility by producing and isolating the NK-EV product (Figure 1). Furthermore, essential product characterization and validation are required before product release is performed, whereby new and original data are presented in this study.
The HFB system was selected for NK-EV production due to its ease of use, reliability, scalability, and GMP compliance7. In reference to the HFB system set-up, the NK cells are injected through the left ECS port and seeded into the bioreactor cartridge (Figure 2). At the same time, the media bottle is connected to the HFB through the side ports, and the media is allowed to flow throughout the system. The NK cells are cultured in serum-free, xeno-free, feeder-free, and antibiotic-free medium, where the media is replaced when the glucose content falls below 50% to maintain and maximize cell health over time. CM is collected daily, processed through differential centrifugations, and kept frozen (-80 °C) until ready for further processing. Afterward, EV isolation is conducted through a combination of differential centrifugations and FPLC-SEC coupled with UF and filtration (Figure 3). This results in a concentrated and sterile NK-EV product with a final volume of approximately 1.0 – 1.5 mL. A representative chromatogram of the FPLC-SEC isolation of NK-EVs is provided (Figure 4). Before FPLC-SEC processing, the NK-EV-rich CM is treated with endonuclease, significantly reducing dsDNA levels, a potential host (NK) cell contaminant7. Thus, the described EV isolation workflow removes cellular debris and RNA/DNA contaminants from the NK-EV product, which is essential for ensuring a low and unwanted immunogenic potential and that the final product is suitable for downstream studies.

Figure 4: NK-EV isolation chromatogram generated during Fast Protein Liquid Chromatography size Exclusion. The blue line represents the absorbance (mAU; maximum reading of 2000 mAU), the red line represents the conductivity, the red text represents the run log, and the gray shaded area represents the fractionated NK-EVs (denoted by fractions T2 – T7). Please click here to view a larger version of this figure.
Following isolation, basic NK-EV characterization and quality assurance testing are used to evaluate if the NK-EV product can be released for further downstream experimentation. NK-EV product particle size range and concentration are measured using nanoparticle tracking analysis (NTA), with sizes ranging from 76.30 – 174.30 nm in diameter (D10 of 78.38 ± 2.07 nm, D50 of 106.72 ± 2.43 nm, and D90 of 169.80 ± 4.17 nm) and an average concentration of 1.39 x 1012 EVs/mL (Figure 5A-B). Additionally, fluorometer quantification showed a protein and dsDNA concentration of 298.90 ± 66.62 mg/mL and 225.60 ± 37.7 ng/mL for the final product, respectively (Figure 5C-D). This corresponds to an average ratio of 5.06 x 106 EV/µg of protein and 6.16 x 1012 EV/µg of DNA. Microbial and mycoplasma testing both returned negative results (data not shown). These results are consistent with the characterization of NK-EVs from previous work7. The earlier publication7 also provides a further in-depth characterization of the NK-EV products following the MISEV guidelines (i.e., TEM, western blot, endotoxin level, viral entities, and flow cytometry for surface antigens and cytokines).
Lastly, the NK-EV product's functionality (i.e., cytotoxicity against cancer cells) was assessed using a validated highly sensitive resazurin-based cell viability assay following NK-EV treatment against leukemic cell line K5627,20. K562 cell treatment with NK-EVs for 3 h produced a dose-dependent effect on cell viability, corresponding to an EC50 of 9.33 x 109 EVs/mL (i.e., the dosage that corresponds to the killing of 50% of the cell population; Figure 6A-B). Thus, following the outlined product release criteria, the NK-EV product is deemed suitable for further experimentation.

Figure 5: Purified NK-EV product characterization. (A) NK-EV product size distribution measured by NTA, shown as mean from 5 independent experiments, each with 10 technical replicates (5 video captures x 2 dilutions). (B) NK-EV particle product concentration (particles/mL) measured by NTA, presented as mean ± SD from 5 independent experiments, each with technical duplicates. (C) NK-EV product protein concentration (mg/mL) was measured by using a fluorometer, presented as mean ± SD from 5 independent experiments, each with technical triplicates. (D) NK-EV product dsDNA concentration (ng/mL) measured by using a fluorometer, presented as mean ± SD from 5 independent experiments, each with technical triplicates. Please click here to view a larger version of this figure.
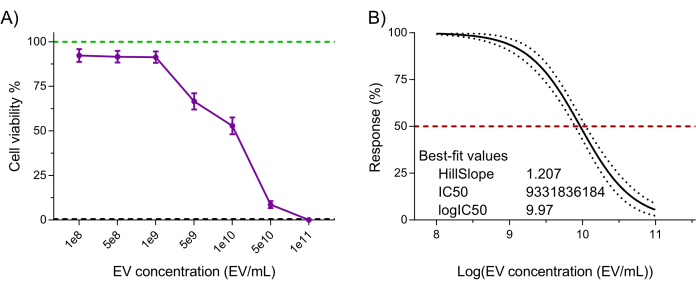
Figure 6: Purified NK-EV product functional validation. NK-EVs demonstrate a dose-dependent cytotoxicity against human K562 leukemia cells treated at various NK-EV concentrations for 3 hours using a highly sensitivity resazurin-based cell viability assay. (A) Normalized assay readouts (green line represents the untreated K562 leukemia cell control, and the black dashed line represents lysed K562 leukemia dead cell control; detergent-treated). Data are shown as mean ± SEM from 11 independent experiments with technical triplicates. (B) EC50 curve analysis with a variable slope for NK-EV treatment with 95% confidence interval/prediction bands (red dashed line represents 50% response). Please click here to view a larger version of this figure.
