Easy Access to Aliphatic Sulfonamides using Sulfamoyl Chlorides Under Visible Light Activation
Özet
Presented here is a protocol for the easy synthesis of aliphatic sulfonamides using sulfamoyl chlorides, (TMS)3SiH and Eosin Y under blue-light irradiation.
Abstract
Sulfonamides are prevalent motifs in marketed drugs and natural products. Their synthesis represents a great interest to the pharmaceutical industry, due to their unique biological properties. Recently, several methods for the synthesis of aryl sulfonamides have been developed, but little effort has focused on developing one-step methodologies to access sulfonamides flanked by two alkyl groups. This protocol describes a practical and facile method for the net hydrosulfamoylation of electron-deficient alkenes using sulfamoyl chlorides as radical precursors under blue-light activation. This practical and cost-effective methodology is performed in the presence of the metal-free photocatalyst Eosin Y and uses light as a clean and traceless energy source. The procedure is scalable, displays a broad functional group tolerance, and can be applied for late-stage functionalization. All reagents used in this protocol are commercially available. Simple reaction set-up, the absence of work-up and easy purification, demonstrate the convenience of this protocol. The reaction is best applied to electron-deficient alkenes.
Introduction
Over the recent decades, sulfonamides featured in a broad range of biologically active molecules and are common motifs in pharmaceuticals and agrochemicals1,2. Initially employed for antibacterial purposes3,4, the application of this motif in drug discovery has been extended to numerous diseases including cancer, CNS disorders, diabetes, dementia and HIV5,6,7,8,9,10,11. Sulfonamides stand out as metabolically stable bioisosteres of carboxylic acids and carboxamides, with the N-H pKa being tunable by varying substitution patterns12,13,14,15.
Traditionally, sulfonamides are synthesized by substitution of a sulfonyl chloride with an amine16,17. The synthesis of sulfonyl chlorides often relies on a multi-step procedure employing harsh conditions, such as strong oxidants. Whilst milder one-step protocols for the installation of sulfonyl chloride intermediates have been developed18,19, the design of a single-step transformation to access sulfonamides is highly desirable.
In the last decades, powerful strategies have been developed for the synthesis of (hetero)aryl sulfonamides, using transition metals, photoredox catalysis or organic catalysts20,21,22,23,24,25,26,27,28,29,30,31,32,33,34. Nevertheless, the one-step synthesis of aliphatic analogues remains underexplored35,36,37,38,39,40. A notable exception is the electrochemical oxidative coupling of amines and thiols, reported by Noël and co-workers41. We were interested in a complementary late-stage functionalization strategy, allowing the direct attachment of commercially available sulfamoyl chlorides onto inexpensive olefins to afford products of net hydrosulfamoylation under visible light activation. Specifically, this process requires an in situ generated sulfamoyl radical, and a suitable hydrogen atom donor.
Preliminary studies indicated that the direct single electron reduction of N,N-dimethylsulfamoyl chloride (Ered = -1.59 V versus saturated calomel electrode (SCE) in MeCN)42 is more challenging than for methanesulfonyl chloride (Ered = -1.30 V versus SCE in MeCN)43, an observation encouraging the identification of an alternative mode of activation to generate sulfamoyl radicals. Inspired by Chatgilialoglu’s work in 198844, we believed that tris(trimethylsilyl)silane can act both as a silyl radical source capable of activating sulfamoyl chlorides, and as the hydrogen atom donor. Blue light irradiation is essential for this reaction to proceed, while Eosin Y is beneficial but not essential.
This practical and cost-effective one-step method tolerates numerous functional groups, thereby allowing access to a broad range of novel alkylsulfonamides including complex sulfonamide-containing cyclobutyl-spirooxindoles that are all valuable building blocks for drug discovery. As part of the challenges faced by industries aiming at avoiding operationally complex, over-engineered, and costly processes, this transformation is not sensitive to oxygen or moisture, uses a metal free photocatalyst, and is operationally simple. Furthermore, the use of blue light as an initiator for this chemical transformation makes this protocol green and sustainable.
Protocol
CAUTION: All chemicals used in this protocol must be handled with care. Please carefully read the material safety data sheets (MSDS) of solvents and reagents used in this protocol. (TMS)3SiH, dimethylsulfamoyl chloride, MeCN, EtOAc and silica have been shown to be toxic, corrosive, irritant, cancerogenic and flammable. Standard lab safety measures are relevant for the handling of those chemicals. All manipulations must be performed in a ventilated laboratory fume hood and the use of appropriate personal protective equipment (PPE), including lab coat, safety glasses, and nitrile gloves is compulsory.
1. Hydrosulfamoylation of electron-deficient alkenes
- Add a magnetic stir bar to a 7 mL vial.
- Weigh out 73.5 mg of N-phenylacrylamide (0.50 mmol, 1.0 equiv) and 1.7 mg of photocatalyst Eosin Y (0.0025 mmol, 0.5 mol%) and add both to the same vial.
- Sequentially add 3.0 mL of MeCN, 309 µL of (TMS)3SiH (1.0 mmol, 2.0 equiv) and 134 µL of N,N-dimethylsulfamoyl chloride (1.25 mmol, 2.5 equiv) with a syringe. Cap the vial with a screw cap.
- Place the vial in the photobox equipped with an 18 W blue LED lamp (λ = 450 nm) and a fan.
- Stir the emulsion vigorously at 1,000 rpm for 4 h.
2. Monitoring of the starting material conversion by thin-layer chromatography (TLC)
- Dissolve 1 mg of N-phenylacrylamide in 1 mL of dichloromethane (DCM). Sample this solution on the TLC plate (left and middle spot).
- Sample a 50 μL aliquot of the reaction mixture and transfer it to a 1.5 mL vial containing 50 μL of DCM. Sample this solution on the TLC plate (middle and right spot).
- Add a solvent mixture of pentane and ethyl acetate (eluent: 80/20 pentane/ethyl acetate) to a TLC chamber.
- Run the TLC plate in the chamber until the solvent front is at 0.5 cm distance of the top of the plate.
- Remove the plate from the chamber, dry it under air and expose the plate to UV light (λ = 254 nm) under a lamp (Rf values: Starting material = 0.4; Product = 0.2).
3. Workup and purification
- Transfer the reaction mixture to a 25 mL round-bottom flask and concentrate the mixture under reduced pressure using a rotary evaporator (150 rpm; until 20 mbar) equipped with a water bath, heated to 40 °C to obtain a crude oil.
- Condition a silica column (pore size 60 Å, 230–400 mesh particle size, 12 g) by passing 60 mL of pentane through the column via a syringe.
- Dilute the crude oil in 2 mL of DCM and transfer the solution onto the column.
- Run a gradient elution on the automated column (EtOAc in pentane 0/100 to 100/0 over 20 min) and monitor by UV-VIS (254 nm) to elute the compounds.
- Collect the fractions in test tubes and monitor the collected fractions by TLC (see section 2).
- Sample aliquots of the collected fractions on a TLC plate.
- Run the TLC plate in the chamber until the solvent front has almost reached the top of the plate and compare the Rf values (see step 2.5).
- Collect the desired fractions as determined by TLC analysis and concentrate the solution under reduced pressure on a rotary evaporator (150 rpm; less than 20 mbar) equipped with a water bath heated to 40 °C.
- Dissolve 5 mg of the product in 0.6 mL CDCl3 and add this solution to a nuclear magnetic resonance spectroscopy (NMR) tube.
- Run a 1H NMR and a 13C NMR and compare the spectra with the information listed below.
Representative Results
The sequence produced the desired hydrosulfamoylated product with 83% yield (106 mg, 0.41 mmol) as an off-white solid. The structure and purity can be assessed by 1H and 13C NMR spectra (Figure 1, Figure 2). More specifically, in the 1H and 13C NMR, disappearance of two characteristic alkene peaks and appearance of two aliphatic peaks, are characteristic for the addition of N,N-dimethylsulfamoyl chloride to the alkene. High-resolution mass spectrometry (HRMS) of the product also confirmed the formation of the desired product.
3-(N,N-dimethylsulfamoyl)-N-phenylpropanamide
1H NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.51 (d, J = 7.6 Hz, 2H), 7.29 (dd, J = 7.9 Hz, 2H), 7.09 (dd, J = 7.4 Hz, 1H), 3.35 (t, J = 7.5 Hz, 2H), 2.91 (t, J = 7.5 Hz, 2H), 2.87 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 167.9, 137.9, 129.1, 124.6, 120.0, 43.5, 37.5, 30.5; HRMS (ESI-TOF) calculated for C11H15O3N232S [M-H]–: 255.0809; found 255.0806; IR (neat) 1681, 1619, 1551, 1491, 1443, 1336, 1314, 1258, 1186, 1147, 952, 760, 742, 684; m.p.: 120‒122 °C.
A wide range of novel aliphatic sulfonamides can be prepared using this methodology in good to high yields42. Each compound has been fully characterized by 1H, 13C NMR, as well as HRMS, IR and melting point42. Note that vigorous stirring is required, due to the use of (TMS)3SiH, which is not miscible in MeCN. Depending on the substrate, the completion of the reaction could be monitored visually as at this point, the mixture becomes homogenous. A color change was observed upon addition of N,N-dimethylsulfamoyl chloride. No degradation of the product was observed when the reaction time was extended to 72 h.
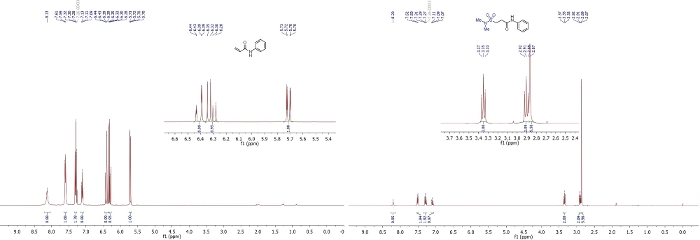
Figure 1: 1H NMR spectra of 1a and 3a (CDCl3, 400 MHz).
This figure has been modified from Gouverneur and co-workers42. Please click here to view a larger version of this figure.
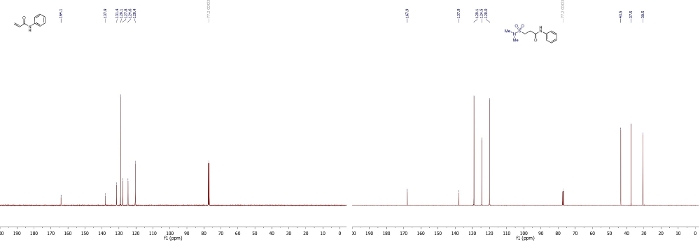
Figure 2: 13C NMR spectra of 1a and 3a (CDCl3, 101 MHz).
This figure has been modified from Gouverneur and co-workers42. Please click here to view a larger version of this figure.
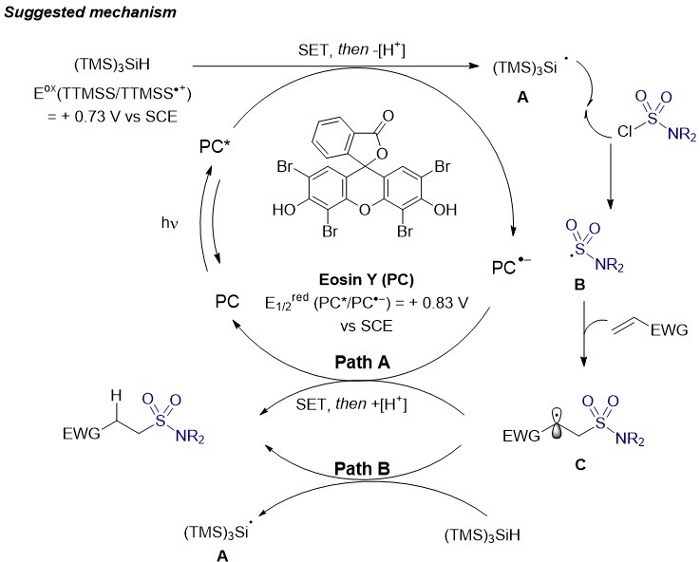
Figure 3: Suggested mechanism for the hydrosulfamoylation of alkenes.
This figure has been modified from Gouverneur and co-workers42. Please click here to view a larger version of this figure.
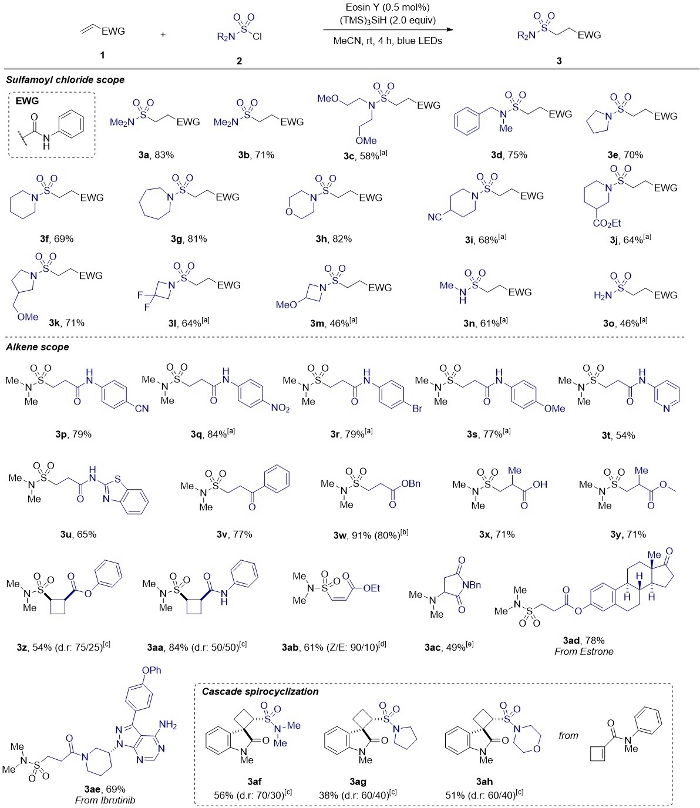
Figure 4: Substrate scope of sulfamoyl chlorides and alkenes.
Reaction conditions: alkene 1 (0.5 mmol), sulfamoyl chloride 2 (1.25 mmol), (TMS)3SiH (1.0 mmol), Eosin Y (0.5 mol%), MeCN (3.0 mL), blue LED irradiation (λmax = 470 nm), room temperature. [a] 16 h reaction time. [b] Scale-up experiment performed on 30.8 mmol (5.0 g) of benzyl acrylate. [c] The diastereomers were separated by silica flash column chromatography. [d] The minor isomer was not isolated. [e] Only traces of the hydrosulfamoylated product was observed. This figure has been modified from Gouverneur and co-workers42. Please click here to view a larger version of this figure.
Discussion
This operationally simple protocol uses commercially available substrates. Nitrogen atmosphere as well as strict water-free conditions are not required for the reaction to proceed in high yields, demonstrating the ease of this protocol. These reactions are often complete within 4 h at room temperature, although some less reactive sulfamoyl chlorides required additional time.
The absence of work-up and the ease of the purification step by silica column chromatography, make this protocol operationally and economically attractive. Interestingly, a fluctuation of the temperature (depending on the distance between the vial and the lamp) did not impact the outcome of the reaction. We noticed that a high purity of the sulfamoyl chlorides was crucial for this transformation to proceed in good yield.
Aiming at broadening the scope, the reactivity of the sulfamoyl radical was investigated towards a range of alkenes of different electronic profiles. As shown previously, the sulfamoyl radical can be added efficiently to electron-deficient alkenes; nevertheless, no conversion was observed with styrenes and unactivated alkenes. Current investigations for the compatibility of these substrates under our reaction conditions is ongoing. Furthermore, sulfonyl chlorides have shown to be reactive under similar reaction conditions45.
A plausible mechanism of the hydrosulfamoylation of electron-deficient alkenes is depicted in Figure 3. Upon irradiation with light, the generated excited triplet state Eosin Y* [E1/2red (PC*/PC•–) = + 0.83 V versus saturated calomel electrode (SCE)] should readily oxidize tris(trimethylsilyl)silane (TTMSS) [Eox (TTMSS/TTMSS•+) = + 0.73 V versus SCE] via single-electron transfer (SET). Upon loss of a proton, silyl radical A is generated, which subsequently abstracts a chlorine-atom from the sulfamoyl chloride to generate sulfamoyl radical B. The latter radical B undergoes regioselective Giese addition to the alkene, whereby the C-centered radical C is formed. The desired hydrosulfamoylated product is finally obtained upon single-electron reduction and protonation (Path A). In this event, the photocatalyst returns to its native oxidation state. A chain propagation reaction mechanism, involving a direct H-atom abstraction from TTMSS is also viable (Path B).
This protocol tolerates a wide range of functional groups, such as esters, amides, carboxylic acids, amines, ethers, halides, nitro and nitriles (Figure 4: 3a‒3ah). The successful introduction of primary, secondary as well as tertiary sulfonamides allowed access to a broad range of novel alkylsulfonamides (3a‒3o). Linear terminal alkenes (3p‒3w), gem-disubstituted alkenes (3x,y), and a representative electron-deficient alkyne (3ab) are all suitable substrates. Cyclobutenes respond well to hydrosulfamoylation generating highly desirable 1,2-disubstituted sulfonamide-containing cyclobutanes (3z,3aa) or cyclobutyl-spirooxindoles (3af‒3ah), all valuable building blocks for drug discovery. The diastereoisomers formed, are easily separable by flash column chromatography. Late-stage functionalization of biologically active molecules was also successful (3ad, 3ae). The scalability of this transformation was demonstrated, providing a short and safe synthetic route to access 3w. We noted competitive desulfonylation with N-benzylmaleimide (3ac).
As an economic and efficient protocol, this protocol could have practical significance for the introduction of aliphatic sulfonamides in complex natural products and biologically active molecules, in both academic and industrial laboratories.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 721902.
Materials
| Acetonitrile | Sigma Aldrich | 34851 | for HPLC, ≥99.9% |
| Biotage | # | ||
| Black Polypropylene Screw Caps | Fisherbrand | 15394789 | – |
| Blue LED | HepatoChem | P201-18-2 450 nm 18W | – |
| Capillary tube | Sigma Aldrich | Z114960 | volume 5-25 µL |
| Eosin Y | Sigma Aldrich | E4009 | Dye content ~99 % |
| EtOAc | Sigma Aldrich | 34858 | for HPLC, ≥99.7% |
| GraceResolv LOK flash cartridge | Grace | 5171343 | |
| Magnetic stirring bar | Biotage | 355543 | – |
| N,N-Dimethylsulfamoyl chloride | Sigma Aldrich | D186252 | – |
| N-Phenylacrylamide | Homemade | – | – |
| Pentane | Sigma Aldrich | 34956 | for HPLC, ≥99.0% |
| Photoredox Box | HepatoChem | HCK1006-01-016 | – |
| TLC Silica gel 60 F254 | Merck | 105554 | aluminium sheets 20 x 20 cm |
| Tris(trimethylsilyl)silane | Combi-Blocks | QF-2110 | – |
| Vial holder | HepatoChem | HCK1006-01-020 | – |
| Vial screw glass 7ml | Samco | T101/V3 | – |
Referanslar
- Feng, M., Tang, B., Liang, S. H., Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Current Topics in Medicinal Chemistry. 16, 1200-1216 (2016).
- Drews, J. Drug discovery: a historical perspective. Science. 287, 1960-1964 (2000).
- Wainwright, M., Kristiansen, J. E. On the 75th anniversary of Prontosil. Dyes Pigments. 88, 231-234 (2011).
- Zaffiri, L., Gardner, J., Toledo-Pereyra, L. H. History of antibiotics. From Salvarsan to Cephalosporins. Journal of Investigative Surgery. 25, 67-77 (2012).
- Casini, A., Scozzafava, A., Mastrolorenzo, A., Supuran, C. T. Sulfonamides and Sulfonylated Derivatives as Anticancer Agents. Current Cancer Drug Targets. 2, 55-75 (2002).
- Shah, S. A., Rivera, G., Ashfaq, M. Recent Advances in Medicinal Chemistry of Sulfonamides. Rational Design as Anti-Tumoral, Anti-Bacterial and Anti-Inflammatory Agents. Mini-Reviews in Medicinal Chemistry. 13, 70-86 (2013).
- Smith, B. R., Eastman, C. M., Njardarson, J. T. Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. Journal of Medicinal Chemistry. 57, 9764-9773 (2014).
- Ilardi, E. A., Vitaku, E., Njardarson, J. T. Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals to Reveal Opportunities for Drug Design and Discovery. Journal of Medicinal Chemistry. 57, 2832-2842 (2014).
- Bag, S., et al. Sulfonamides as multifunctional agents for Alzheimer’s disease. Bioorganic & Medicinal Chemistry Letters. 25, 626-630 (2015).
- Gao, H. D., Liu, P., Yang, Y., Gao, F. Sulfonamide-1,3,5-triazine–thiazoles: discovery of a novel class of antidiabetic agents via inhibition of DPP-4. RSC Advances. 6, 83438-83447 (2016).
- Apaydın, S., Török, M. Sulfonamide derivatives as multi-target agents for complex diseases. Bioorganic & Medicinal Chemistry Letters. 29, 2042-2050 (2019).
- Pinter, T., Jana, S., Courtemanche, R. J. M., Hof, F. Recognition Properties of Carboxylic Acid Bioisosteres: Anion Binding by Tetrazoles, Aryl Sulfonamides, and Acyl Sulfonamides on a Calix[4]arene Scaffold. The Journal of Organic Chemistry. 76, 3733-3741 (2011).
- Ballatore, C., Huryn, D. M., Smith, A. B. Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem. 8, 385-395 (2013).
- Lassalas, P., et al. Structure Property Relationships of Carboxylic Acid Isosteres. Journal of Medicinal Chemistry. 59, 3183-3203 (2016).
- Şanli, N., Şanli, S., Özkan, G., Denizlic, A. Determination of pKa values of some sulfonamides by LC and LC-PDA methods in acetonitrile-water binary mixtures. Journal of the Brazilian Chemical Society. 21, 1952-1960 (2010).
- Bahrami, K., Khodaei, M. M., Soheilizad, M. Direct Conversion of Thiols to Sulfonyl Chlorides and Sulfonamides. The Journal of Organic Chemistry. 74, 9287-9291 (2009).
- Veisi, H., Ghorbani-Vaghei, R., Hemmati, S., Mahmoodi, J. Convenient One-Pot Synthesis of Sulfonamides and Sulfonyl Azides from Thiols Using N-Chlorosuccinimide. Synlett. 16, 2315-2320 (2011).
- Rawner, T., Knorn, M., Lutsker, E., Hossain, A., Reiser, O. Synthesis of Trifluoromethylated Sultones from Alkenols Using a Copper Photoredox Catalyst. The Journal of Organic Chemistry. 81, 7139-7147 (2016).
- Bagal, D. B., et al. Trifluoromethylchlorosulfonylation of Alkenes:Evidence for an Inner-Sphere Mechanism by a Copper Phenanthroline Photoredox Catalyst. Angewandte Chemie International Edition. 54, 6999 (2015).
- Yin, J., Buchwald, S. L. Palladium-Catalyzed Intermolecular Coupling of Aryl Halides and Amides. Organic Letters. 2, 1101-1104 (2000).
- Burton, G., Cao, P., Li, G., Rivero, R. Palladium-Catalyzed Intermolecular Coupling of Aryl Chlorides and Sulfonamides under Microwave Irradiation. Organic Letters. 5, 4373-4376 (2003).
- Shaabani, A., Soleimani, E., Rezayan, A. H. A novel approach for the synthesis of alkyl and aryl sulfonamides. Tetrahedron Letters. 48, 2185-2188 (2007).
- Baffoe, J., Hoe, M. Y., Touré, B. B. Copper-Mediated N-Heteroarylation of Primary Sulfonamides: Synthesis of Mono-N-heteroaryl Sulfonamides. Organic Letters. 12, 1532-1535 (2010).
- DeBergh, J. R., Niljianskul, N., Buchwald, S. L. Synthesis of Aryl Sulfonamides via Palladium-Catalyzed Chlorosulfonylation of Arylboronic Acids. Journal of the American Chemical Society. 135, 10638-10641 (2013).
- Yang, B., et al. Synthesis of N-arylsulfonamides through a Pd-catalyzed reduction coupling reaction of nitroarenes with sodium arylsulfinates. Organic & Biomolecular Chemistry. 16, 8150-8154 (2018).
- Chen, Y., Murray, P. R. D., Davies, A. T., Willis, M. C. Direct Copper-Catalyzed Three-Component Synthesis of Sulfonamides. Journal of the American Chemical Society. 140, 8781-8787 (2018).
- Kim, T., McCarver, S. J., Lee, C., MacMillan, D. W. C. Sulfonamidation of Aryl and Heteroaryl Halides through Photosensitized Nickel Catalysis. Angewandte Chemie International Edition. 57, 3488-3492 (2018).
- Chan, W. Y., Berthelette, C. A mild, efficient method for the synthesis of aromatic and aliphatic sulfonamides. Tetrahedron Letters. 43, 4537-4540 (2002).
- Burrows, J. N., Tucker, H. 1-Sulphonyl piperidine derivatives. , (2004).
- Burns, D. M., Yao, W., He, C. Hydroxamic acid derivatives as metalloprotease inhibitors. , (2005).
- Cumming, J. N., Gilbert, E. J., Stamford, A. W. C5-c6 oxacyclic-fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use. , (2012).
- Joyard, Y., Papamicaël, C., Bohn, P., Bischoff, L. Synthesis of Sulfonic Acid Derivatives by Oxidative Deprotection of Thiols Using tert-Butyl Hypochlorite. Organic Letters. 15, 2294-2297 (2013).
- Shavnya, A., Coffey, S. B., Hesp, K. D., Ross, S. C., Tsai, A. S. Reaction of Alkyl Halides with Rongalite: One-Pot and Telescoped Syntheses of Aliphatic Sulfonamides, Sulfonyl Fluorides, and Unsymmetrical Sulfones. Organic Letters. 18, 5848-5851 (2016).
- Wang, M., Fan, Q., Jiang, X. Metal-free construction of primary sulfonamides through three diverse salts. Green Chemistry. 20, 5469-5473 (2018).
- Wallentin, C. J., Nguyen, J. D., Finkbeiner, P., Stephenson, C. R. J. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. Journal of the American Chemical Society. 134, 8875-8884 (2012).
- Jiang, H., Cheng, Y., Zhang, Y., Yu, S. Sulfonation and Trifluoromethylation of Enol Acetates with Sulfonyl Chlorides Using Visible-Light Photoredox Catalysis. European Journal of Organic Chemistry. 24, 5485-5492 (2013).
- Liu, X., Cong, T., Liu, P., Sun, P. Visible light-promoted synthesis of 4-(sulfonylmethyl)isoquinoline-1,3(2H,4H)-diones via a tandem radical cyclization and sulfonylation reaction. Organic & Biomolecular Chemistry. 14, 9416-9422 (2016).
- Pagire, S. K., Paria, S., Reiser, O. Synthesis of β-Hydroxysulfones from Sulfonyl Chlorides and Alkenes Utilizing Visible Light Photocatalytic Sequences. Organic Letters. 18, 2106-2109 (2016).
- Alkan-Zambada, M., Hu, X. Cu-Catalyzed Photoredox Chlorosulfonation of Alkenes and Alkynes. The Journal of Organic Chemistry. 84, 4525-4533 (2019).
- Luo, Q., Mao, R., Zhu, Y., Wang, Y. Photoredox-Catalyzed Generation of Sulfamyl Radicals: Sulfonamidation of Enol Silyl Ether with Chlorosulfonamide. The Journal of Organic Chemistry. 84, 13897-13907 (2019).
- Laudadio, G., et al. Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. Journal of the American Chemical Society. 141, 5664-5668 (2019).
- Hell, S. M., et al. Silyl Radical-Mediated Activation of Sulfamoyl Chlorides Enables Direct Access to Aliphatic Sulfonamides from Alkenes. Journal of the American Chemical Society. 142, (2020).
- Ignat’ev, N., Kucherina, A., Sartori, P. Comparative Electrochemical Fluorination of Ethanesulfonyl Chloride and Fluoride. Acta Chemica Scandinavica. 53, 1110-1116 (1999).
- Chatgilialoglu, C., Griller, D., Rossini, S. Amino- and Alkoxysulfonyl Radicals. The Journal of Organic Chemistry. 54, 2734-2737 (1989).
- Hell, S. M., et al. Hydrosulfonylation of Alkenes with Sulfonyl Chlorides under Visible Light Activation. Angewandte Chemie International Edition. 59, 11620-11626 (2020).

.