In this section, representative results are provided for the protocol used for examination of a single subunit E3 ubiquitin ligase RHA1B that has a PROSITE-predicted RING-H2 type domain (132–176 amino acids)10. As shown in Figure 1, in order to obtain an E3-deficient mutant protein, at least one of the eight conserved Cs or Hs in the RING domain (Figure 1A) needs to be mutagenized (Figure 1B). Thus, as a first step, two mutant versions of RHA1B, RHA1BC135S (a substitution of Cys by Ser in the conserved C3 of RING domain) and RHA1BK146R (a substitution of Lys by Arg in the only Lys present in RHA1B) were generated. Although single subunit E3 ligases mediate ubiquitin transfer from ubiquitin harboring E2 to the substrate rather than directly interacting with ubiquitin, self-ubiquitination of E3 at Lys might be required for its maximal enzymatic activity.
The Western blotting results in Figure 2A show a typical positive in vitro ubiquitination assay outcome, with a multibanding smear starting at the molecular weight of the tested protein (e. g., MPB-fused RHA1B ~100 kDa) and progressing upwards. The anti-HA antibody recognized HA-tagged Ub incorporated into the poly-ubiquitination chain of different lengths, creating this typical ubiquitin-associated ladder-like smear. To validate the positive results, Figure 2A also presents all important negative controls missing individual components (E1, E2, Ub, or MBP-RHA1B) or using MBP as control and lacking the smeared ubiquitination signal. Furthermore, the Coomassie blue staining of the PVDF membrane showed equal loading of MBP-RHA1B or MBP in all controls.
Figure 2B shows how in vitro ubiquitination results varied depending on the specific E2/E3 combination. In this example, 11 different E2s representing 10 different E2 families were tested. The detected ubiquitination activity ranged from no signal (no smear) to a multibanding smear starting at different molecular weights, indicating different ubiquitination patterns.
Figure 3 shows ubiquitination assay outcomes for RING- and K-mutant versions of tested protein. Lack of enzymatic activity for RHA1BC135S is supported by its inability to either generate a multibanding smear in vitro (Figure 3A) or promote poly-ubiquitination signal in planta (Figure 3B). It is notable that overexpression of HA-tagged Ub in planta on its own gave basal level ubiquitination in all tested samples, including the vector control, in contrast to the strong ubiquitination signal conferred by the enzymatic activity of wild type RHA1B. Furthermore, the analysis on the RHA1BK146R mutant suggests that the K146 residue is also essential for the E3 activity of RHA1B. Although a marginal self-ubiquitination signal was detected in vitro (Figure 3A), the in planta assay determined the mutant is E3-deficient (Figure 3B, only background ubiquitination signal detected).
After generating and biochemically validating the E3-deficient mutant, functional studies can be designed to determine the E3-associated biological role of the tested RING E3 ubiquitin ligase. In the case of RHA1B, this nematode effector suppresses plant immune signaling, as manifested by suppression of the Gpa2-triggered HR cell death. As presented in Figure 4A, unlike the wild type RHA1B, the RHA1BC135S mutant lacking E3 ligase activity did not interfere with HR cell death. Given that the most common outcome of protein ubiquitination is its proteasome-mediated degradation, mutations residing in the RING domain can also be used to verify an E3-dependent ability to trigger degradation of their direct and/or indirect substrates. Thus, significantly, Western blotting results in Figure 4B confirm that Gpa2 did not accumulate in the presence of wild type RHA1B but RHA1BC135S had no impact on Gpa2 protein stability.
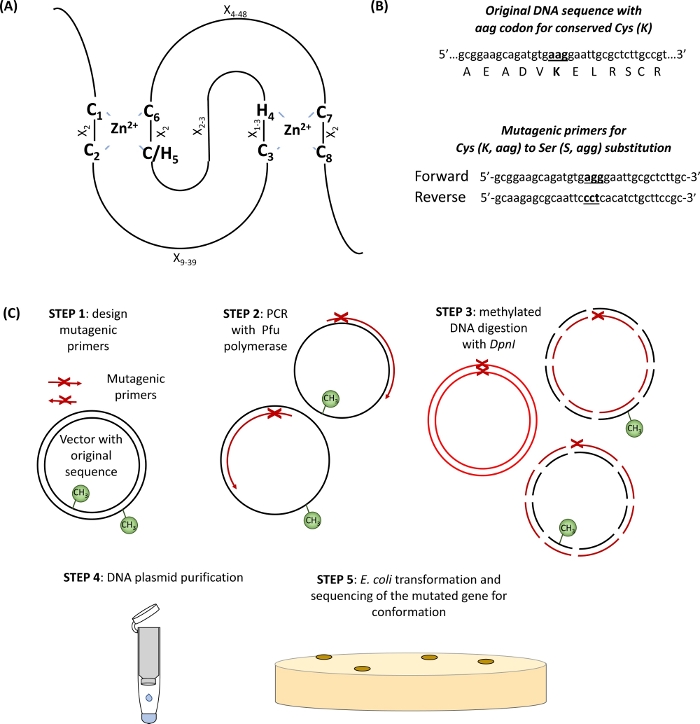
Figure 1: Schematic representation of the principle and steps involved in site-directed mutagenesis. (A) RING-CH/H2 domain with conserved Cys and His amino acids highlighted. (B) An example of mutagenic primers design. (C) Steps of site-directed mutagenesis. Please click here to view a larger version of this figure.
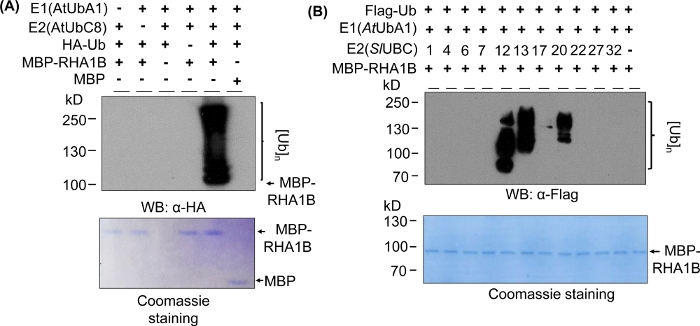
Figure 2: Representative in vitro ubiquitination assay. (A) Top gel shows ubiquitination assay including all negative controls, and bottom gel shows equal loading. (B) The range of expected results depending on E2 enzymes. This figure has been modified from Kud et al8. Please click here to view a larger version of this figure.
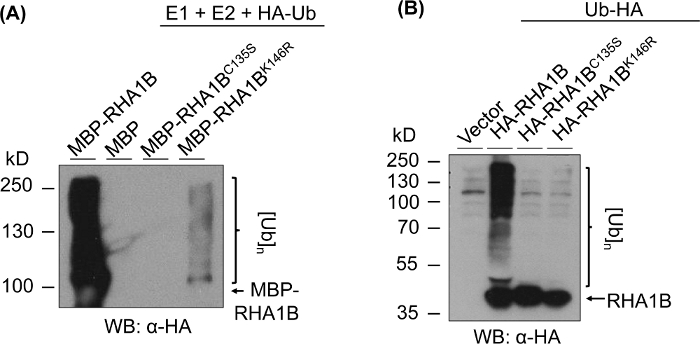
Figure 3: Ubiquitination assay results for RING- and K- mutants (RHA1BC135S and RHA1BK146R). (A) In vitro ubiquitination results for RHA1BC135S and RHA1BK146R. (B) In planta ubiquitination assay results for RHA1BC135S and RHA1BK146R. This figure has been modified from Kud et al8. Please click here to view a larger version of this figure.
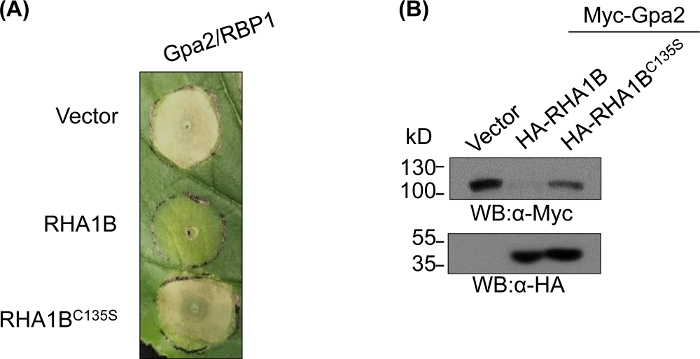
Figure 4: Representative functional study for E3 dependent biological functions. An example of functional studies showing the E3-dependent biological function. (A) E3-dependent HR cell death suppression and (B) degradation of a plant immunoreceptor Gpa2. This figure has been modified from Kud et al8. Please click here to view a larger version of this figure.
| PCR set up | |
| 1 µL | plasmid (~100 ng) |
| 1.5 µL | F mutagenic primer (10 µM) |
| 1.5 µL | R mutagenic primer (10 µM) |
| 1 µL | dNTPs (10 mM) |
| 5 µL | buffer (10x) |
| 1 µL | Ultra Pfu polymerase (2.5 U/µl) |
| 39 µL | ddH2O |
| 50 µL | TOTAL VOLUME |
Table 1: PCR reaction set up
| thermocycler program | |||
| 1 | 95 ˚C | 30 s | |
| 2 | 95 ˚C | 30 s | |
| 3 | 60 ˚C | 30 s | |
| 4 | 72 ˚C | 5 min | repeat 2-4 30 times |
| 5 | 72 ˚C | 5 min | |
Table 2: PCR thermocycler program
| ligation reaction set up for the RHA1B example | ||
| 1.5 µL | pMAL-c2::MBP linearized vector by digestion with BamHI and SalI (60 ng) | |
| 7 µL | RHA1B/RHA1BC135S or RHA1BK146R insert digested with BamHI and SalI (25 ng) | |
| 1 µL | T4 ligase buffer (10x) | |
| 0.5 µL | T4 ligase (400 U/µL) | |
| 10 µL | TOTAL VOLUME | |
Table 3: Ligation reaction set up for the RHA1B example.
