One of the main challenges to attributing biologicals effects to extracellular vesicles (EVs) is the ability to isolate the EVs from the whole culture medium. In this report, we present a method using ultracentrifugation (UC) and size-exclusion chromatography (SEC) which is coupled to the large-scale analysis of protein signatures to validate EV markers and identify bioactive compounds. The macrophage- or microglia-derived EVs were isolated from the conditioned medium after a 24 h or 48 h culture respectively (Figure 1).
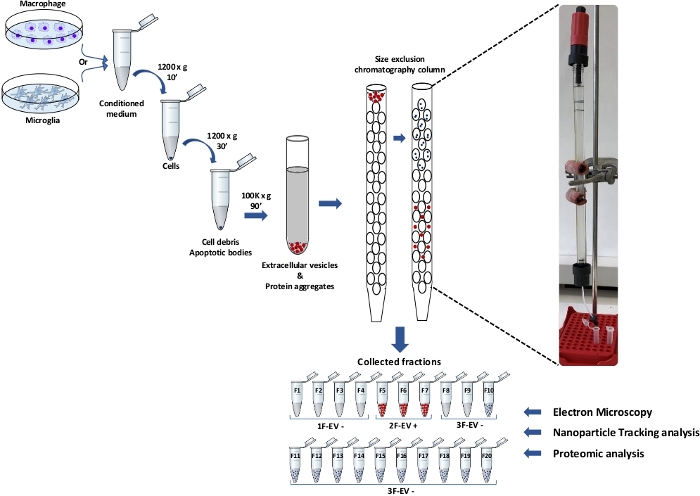
Figure 1: Extracellular Vesicle (EV) collection and isolation strategies. The apoptotic bodies and the cell debris were separated from the microglia- or macrophage-conditioned medium by successive centrifugation steps. From the supernatant, EVs were isolated by ultracentrifugation (UC). The UC pellet containing EVs was loaded onto column and separated by size-exclusion chromatography (SEC) in 20 different eluted fractions. As revealed in the further steps (electron microscopy, nanoparticle tracking analysis and proteomic analysis), the SEC fractions were pooled and organized into 1F-EV-, 2F-EV+ and 3F-EV-. Please click here to view a larger version of this figure.
The method followed successive centrifugation steps: the first to remove the cells and the second to remove cellular debris and apoptotic bodies. Then, the supernatant was transferred to a new tube to perform an ultracentrifugation at 100,000 x g for 90 min. The vesicles were collected together with the protein aggregates in the final UC pellet. An SEC column was used to separate the compounds according to their size and remove the aggregates (Figure 1). In order to confirm the isolation of EVs, each SEC fraction followed a nanoparticle tracking analysis (Figure 2A).
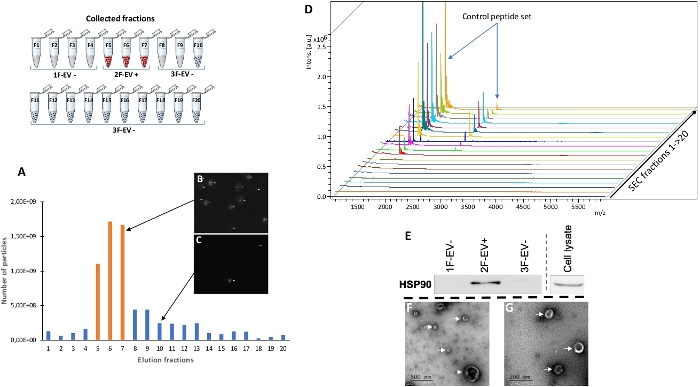
Figure 2: Analysis of the SEC fractions. (A) Nanoparticle tracking analysis (NTA) of SEC fractions. The total number of particles in each SEC fraction is presented with bar charts. The orange bar charts show the three consecutive EV+ fractions (F5–F7). (B, C) Screen captures of NTA chamber show significant differences in particle flow between SEC fractions. (D) Complementary MALDI mass spectrometry analysis. From another UC pellet containing EVs, a standard control peptide set was added before SEC separation in order to validate the performance of the SEC strategy. Each SEC fraction was analyzed with matrix assisted laser desorption ionization – time of flight (MALDI-TOF) to determine standard-positive fractions. In the nine first spectra (fractions 1 to 9), no signal was observed. The detection of these free standards was possible in the following fractions (fractions 10 to 20) confirming the ability of SEC procedure to separate EVs (F5–F7) from soluble components (F10–F20). (E) Western blot analysis of SEC fractions as an EV marker preliminary analysis. The EV+ fractions (F5–F7) were pooled as one sample (2F-VE+) whereas the EV- fractions (F1–F4 and F8–F20 respectively) were pooled in two other samples called 1F-EV- and 3F-EV-. The results showed the presence of a Heat-Shock Protein 90 (HSP90) (EV positive marker) signal in 2F-EV+, and also in cell lysate as positive control, compared to the other fractions (1F-EV- and 3F-EV-). (F, G) Electron microscopy analysis of the EV positive sample (2F-EV+). The observation under two successive magnifications revealed the presence of EVs in a size range around 100 nm (white arrows) and around 400 nm (arrowhead). Please click here to view a larger version of this figure.
The particle number was significantly higher in the fractions 5, 6 and 7 than in the previous (F1–F4) and following ones (F8–F20). It was possible to see a particle flow in these fractions even if a few particle were observed in the following fractions (Figure 2B,C). This separation does not prevent the possibility that fractions other than F5–F7 may contain a small amount of vesicles, or even that EV-rich fractions F5 to F7 may contain molecular contaminants. To at least ensure that the F5–F7 fractions are free of contaminating co-eluting molecules, it was necessary to follow the time-course elution of the different compounds in the SEC procedure. To do so, control peptide standards were added to a similar UC pellet as previously used. This preparation was again separated using the SEC procedure. Then, a matrix assisted laser desorption ionization — time of flight (MALDI) mass spectrometry analysis was performed in order to identify the SEC fractions in which standards were eluted (Figure 2D). Due to the mass range, only ionized products from peptide standards were followed in this analysis. The results showed that these products were detectable in the fractions 10 to 20, demonstrating that any soluble protein cannot be eluted in the same fractions as the EVs (F5–F7). These results confirmed the interest of this approach in the separation of EVs from non-EV components. Even assuming that the F8–F20 fractions could contain a residual fraction of EVs, we decided in the following experiments to combine the EV positive fractions (F5–F7) in a single sample called 2F-EV+. We also combined the other SEC fractions into two samples called 1F-EV- (F1–F4) and 3F-EV- (F8–F20). We grouped in three samples for several reasons. First, the number of SEC fractions would increase the time of molecular analyses but also functional in vitro studies. The EV-positive SEC fractions separately exhibit lower particle numbers and also compromise the detection of molecular compounds. In the same way, it was considered more judicious to group the fractions F8–F20 to concentrate, if necessary, the residual vesicles and check their presence by a preliminary western blot analysis against HSP90, an EV marker. Interestingly, a positive signal for HSP90 was detected in the fraction 2F-EV+, also in the cell lysate as positive control, but not in 1F-EV- and 3F-EV- samples (Figure 2E and Supplementary Figure S1 as control). This analysis confirms the interest of 2F-EV+ only and the correct management of the previous SEC fractions. To affirm EV isolation, an additional experiment using electron microscopy analyzed only the 2F-EV+ sample and allowed the observation of EVs with a typical morphology and size heterogeneity between 100 and 400 nm (Figure 2F,G).
Another key step in the validation was proteomic analysis (Figure 3). We developed a whole mass spectrometry analysis with multiple objectives: (i) confirm the isolation of EVs with the detection of a high number of EV markers in 2F-EV+, (ii) eventually identify contaminant proteins in the EV negative samples (1F-EV- and 3F-EV-) and (iii) characterize the protein contents of EVs supporting their biological activities.
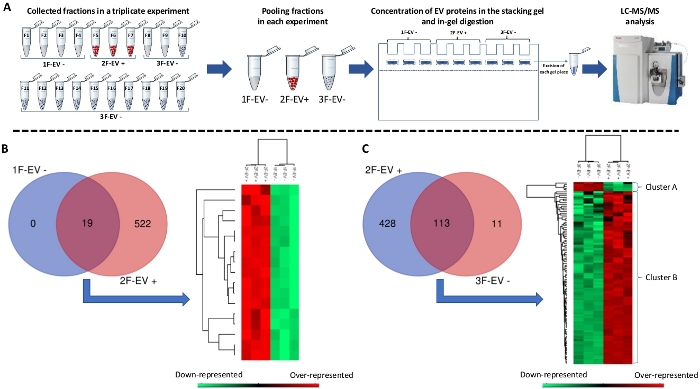
Figure 3: Proteomic analysis of EV-positive and EV-negative samples. (A) After three independent EV isolations from similar microglia cell preparations, the samples 1F-EV-, 2F-EV+ and 3F-EV- were loaded for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and migrated until the stacking gel to concentrate proteins in order to realize their in-gel digestion. The resulting peptides were then analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) to identify the corresponding proteins. (B) Comparison of identified proteins between samples 1F-EV- and 2F-EV+ showing common proteins in both fractions (19) and proteins exclusively detected in the 2F-EV+ fraction (522). The heatmap shows the relative representation where all common proteins between 1F-EV- and 2F-EV+ triplicates were over-represented (red) in the 2F-EV+ samples. (C) Comparison of identified proteins between fractions 2F-EV+ and 3F-EV- showing common proteins between triplicates (113) and exclusively represented proteins (428 in 2F-EV+ and 11 in 3F-EV-). The heatmap shows the relative representation in two clusters. One (cluster A) presents 5 over-represented proteins in 3F-EV- sample and a second (cluster B) presents 108 over-represented proteins in 2F-EV+. Please click here to view a larger version of this figure.
From cell-conditioned media, the UC and SEC procedure led to three samples 1F-EV-, 2F-EV+ and 3F-EV-. The corresponding proteins were extracted and concentrated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After a short migration in the stacking gel, the samples were collected by a band excision, transferred in distinct tubes to be in-gel digested. The products were separated by online reversed-phase chromatography directly coupled to a mass spectrometer for analysis (Figure 3A).
The following results, as an example of the whole procedure, were obtained from microglia-conditioned medium after a 48 h culture. The raw data were submitted to a Rat protein database. The identified proteins were compared between samples 1F-EV- and 2F-EV+ (Figure 3B) and between samples 2F-EV+ and 3F-EV- (Figure 3C). In the first comparison, the Venn diagram showed 19 common proteins in both fractions and 522 proteins exclusively detected in the 2F-EV+ fraction. The analysis using a quantitative proteomics software allowed the identification of the relative representation of the common proteins. The results showed a heatmap where all common proteins between 1F-EV- and 2F-EV+ triplicates were over-represented (red) in the 2F-EV+ samples. In the second comparison between fractions 2F-EV+ and 3F-EV-, 113 common proteins were identified between triplicates. Moreover, 428 proteins were exclusively detected in 2F-EV+ and 11 proteins were exclusively found in 3F-EV-. Following the quantitative analysis, the heatmap representation of the 113 common proteins highlighted two clusters where the cluster A presented five over-represented proteins in 3F-EV- sample and the cluster B presented 108 over-represented proteins in 2F-EV+.
The 428 proteins exclusively represented and the 108 proteins over-represented in 2F-EV+ were submitted to the Exocarta open access database in order to detect EV-associated molecules. The analysis of the top 100 EV markers in Exocarta highlighted the presence of 86 EV-associated proteins in 2F-EV+ (Figure 4A).
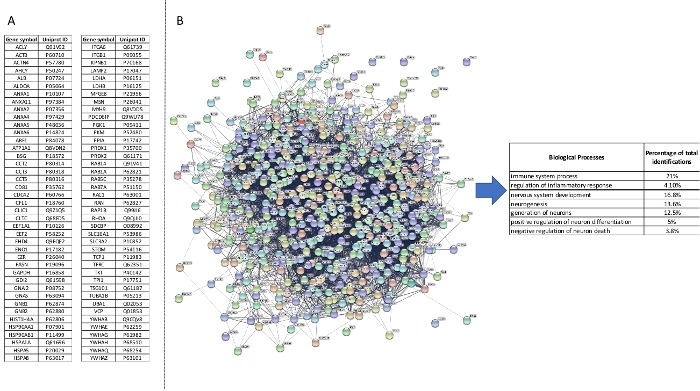
Figure 4: Analysis of proteins from the 2F-EV+ sample. (A) A pool of 536 proteins (428 exclusive and 108 over-represented proteins) was submitted to the Exocarta database in order to detect EV-associated molecules. Molecule symbols of the 86 EV-associated proteins detected in the top 100 EV markers from Exocarta database. (B) The prediction of protein interactions and their association to selected biological processes showed immune and neuroprotective pathways in the 2F-EV+ sample. Please click here to view a larger version of this figure.
Interestingly, the analysis of the 5 common proteins in the cluster A and the 11 proteins exclusively represented in 3F-EV- were not associated to any EV marker (data not shown). Finally, the prediction of protein interactions and their association with selected biological processes showed the presence in the 2F-EV+ fraction of immune mediators (21%) sometimes involved in the control of the inflammatory response (4.1%) (Figure 4B). The EV contents in 2F-EV+ were also associated with the neuronal development (16.8%), neuron differentiation (5%) and the control of neuronal death (3.8%) which is in accordance with the following neurite outgrowth assay.
Thus, the strategy that we selected makes possible the access of all data and allows a prediction of the EV-mediated functions prior to the biological assays that we then performed (Figure 5).
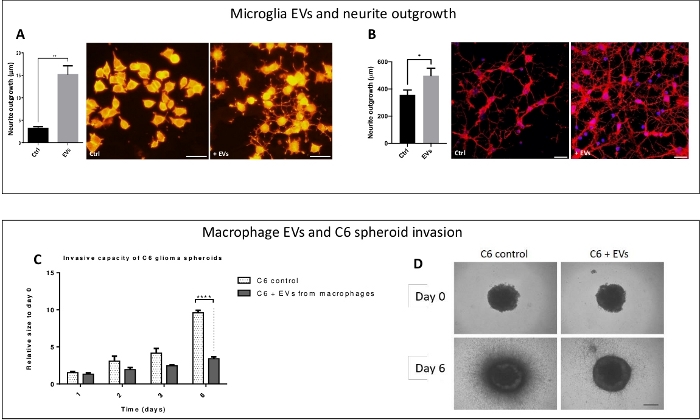
Figure 5: EV-dependent f unctional assays. The effects of microglia-derived EVs were evaluated on neurite outgrowth (upper frame) and the effects of macrophage-derived EVs were evaluated on glioma cell invasion (lower frame). (A, B) The neurite length was measured on PC-12 cells (Panel A was modified from Raffo-Romero et al.16 with permission) or rat primary neurons (B). The results showed a significant outgrowth increase under EVs (+ EVs) compared to control (Ctrl). Scale bar = 20 µm. (C, D) Time course of C6 glioma spheroid invasion into collagen in the presence of rat primary macrophage-derived EVs (C6 + EVs) or vehicle (C6 control). The C6 spheroid 3D invasion into collagen was monitored and quantified up to 6 days. Quantification of tumor cell invasion are shown at 1, 2, 3 or 6 days (C) as observed on representative images (D). Scale bar = 500 µm. For neurite outgrowth assays, the significance was calculated by unpaired Student's t-test (* p < 0.05, ** p < 0.01 and *** p < 0.001). For invasion assay, the significance was calculated by one-way ANOVA followed by Tukey's post hoc test. The error bars indicate relative standard error of three independent experiments. Please click here to view a larger version of this figure.
In this way, the neurotrophic functions of microglia-derived EVs were studied on PC-12 cell line and rat primary neurons. The results showed a positive effect on the neurite outgrowth (Figure 5A,B). The presence of EVs respectively increased about 6 and 1.5 fold the neurite network in length and/or in number in PC-12 and primary neurons compared to a control condition. In another context, we also studied the effects of macrophage-derived EVs on a brain tumor invasion (Figure 5C,D). It was assessed using 3D tumor spheroids embedded in a matrix of collagen. Spheroids generated with C6 rat glioma cells were cultured for 6 days in a collagen matrix containing (or not containing) EVs purified from rat macrophages culture medium. The collagen matrix provides a structure into which tumor cells can invade and spread out of the spheroid. The macrophage-derived EVs impaired the growth and invasion of the glioma spheroids. After a 6 day culture, a 50% decrease of the invasion was observed in EV-treated spheroids compared to the control. This result showed that macrophages can produce EVs with anti-tumoral and/or anti-invasive factors affecting the glioma growth.
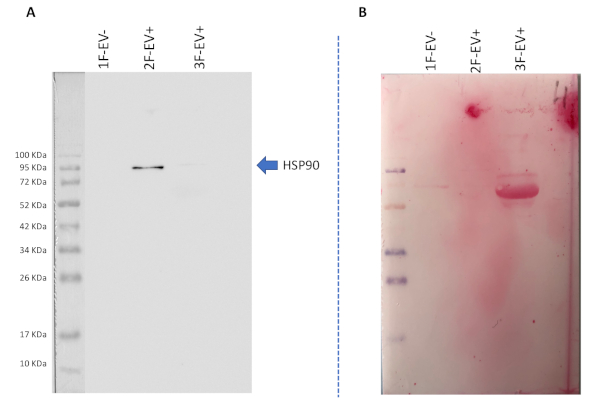
Supplementary Figure S1: Images of membrane used for western-blotting experiment. (A) Original image of the western blot from Figure 2E (against HSP90 EV marker). (B) Ponceau staining image of the same membrane showing the correct loading of protein extracts from 1F-EV-, 2F-EV+ and 3F-EV- samples. Please click here to download this figure.
