In a representative experiment to test the presented protocol we extracted the RNA from 10 different blood samples of five animal species (i.e., 2 individuals per species (goat, sheep, swine, dog, cattle)) (Table 3). RNA yields and quality following extraction can vary widely, in particular due to differences in sample handling and storage. In our representative experiment, we observed RNA concentrations between 43 ng and 543 ng per µL (Table 3). Also, after amplification by RT-PCR, gel analysis of the NPC-1 PCR-products showed various outcomes (Figure 2), with markedly weaker bands for samples BC01 and BC02 (both goat). These differences were most likely caused by differences in sample quality, although differences in PCR efficacy due to differences in primer binding to the NPC1 gene of different species cannot be excluded. However, these differences in yield and/or amplification efficiency did not markedly impact the overall sequencing outcome. Further, an additional non-specific PCR product occurred in sample BC10 (cattle). In contrast to Sanger sequencing, such non-specific products do not negatively influence the results of nanopore sequencing, as these reads are discarded during mapping of the obtained reads to a reference sequence as part of the data analysis.
Prior to each sequencing run, a quality check of the flow cell to be used is strongly recommended, with a minimum requirement of 800 total pores. In our representative experiment, this quality check returned 1,102 pores available for sequencing. Since the data are provided in real time and can be analyzed immediately, the length of a sequencing run can be adjusted for the individual application (i.e., until sufficient sequencing data is produced for the desired analysis). In our experiments, sequencing runs are typically performed overnight, and in the case of our representative experiment we obtained approximately 1.4 million reads during such a 14 h run.
Depending on the type of data analysis to be performed, it can be advisable to process only a subset of the obtained reads. In the case of our representative experiment, a subset of 10,000 reads was selected for further analysis. To this end, the fastq files generated during the sequencing run were further processed in an Ubuntu 18.04 LTS environment, and demultiplexed using flexbar v3.0.3 with parameters optimized for demultiplexing of nanopore sequencing data (barcode-tail-length 300, barcode-error-rate 0.2, barcode-gap-penalty -1)12. After demultiplexing, read mapping and consensus generation can then be done using a number of different tools, but a detailed discussion of the bioinformatics aspect of nanopore sequencing goes beyond the scope of this manuscript. However, in the case of our representative results, read mapping to a reference sequence was performed using Geneious 10.2.3. Of the 10,000 reads analyzed, 5,457 showed a length between 1,750 and 2,000 nucleotides, which matches the expected sizes for the PCR fragments amplified as part of our workflow (1,769 nt, Figure 3). An additional peak in the length distribution of reads was observed between 250 to 500 nucleotides, which can be attributed to unspecific PCR products. Demultiplexing of reads allowed the assignment of 87.6% of the reads to one of the 10 barcodes/samples analyzed (Figure 4). The proportion of demultiplexed reads for each barcode ranged from 3.4% for barcode 1 to 16.9% for barcode 10; however, due to the overall large number of reads this still allowed meaningful consensus calling with a high read depth even for these lower abundance barcode datasets. Indeed, mapping of the sorted reads to a reference sequence of NPC1 resulted in between 31.7% (barcode 2) and 100% (barcode 7 and 8) of reads mapping to the reference, giving a read depth of more than 90 reads at any position for each sample. This is then more than adequate to allow confident consensus base-calling with a negligible error rate.
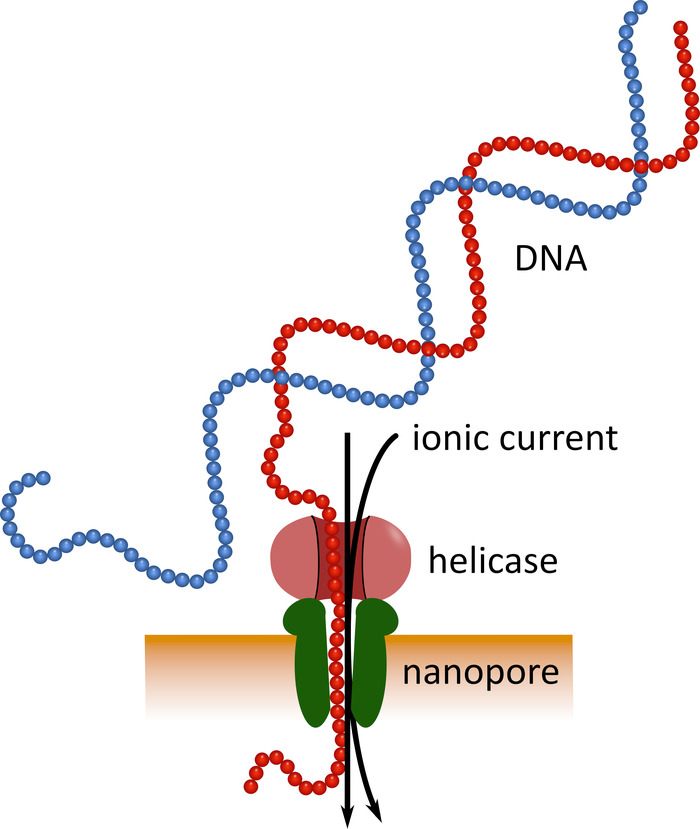
Figure 1: Schematic representation of DNA sequencing using nanopore technology. A single-stranded DNA molecule passes through a nanopore embedded in an electrically resistant membrane, with a helicase regulating the transition speed. An ionic current simultaneously passes through the pore and is continuously measured. Modulations of the current caused by the nucleotides present in the pore are detected and computationally back-translated into the nucleotide sequence of the DNA strand. Please click here to view a larger version of this figure.
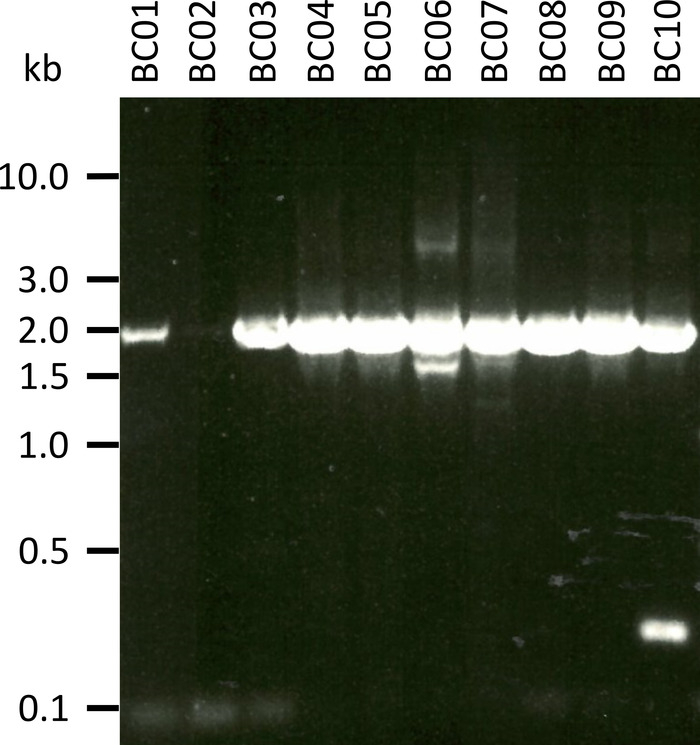
Figure 2: Amplification of PCR products of Niemann-Pick C1 from mRNA. mRNA was isolated from goat (BC01 and 02), sheep (BC03 and 04), swine (BC05 and 06), dog (BC07 and 08), and cattle (BC09 and 10). Nested PCR products were separated in a 0.8% agarose gel in 1x TAE buffer (prepared from 50x TAE buffer: 242.28 g of Tris base, 57.1 mL of glacial acetic acid, 100 mL of 0.5 M EDTA, dH2O to 1 L, pH adjusted to 8.0) for 45 min at 100 V and stained with Sybr Safe. Please click here to view a larger version of this figure.
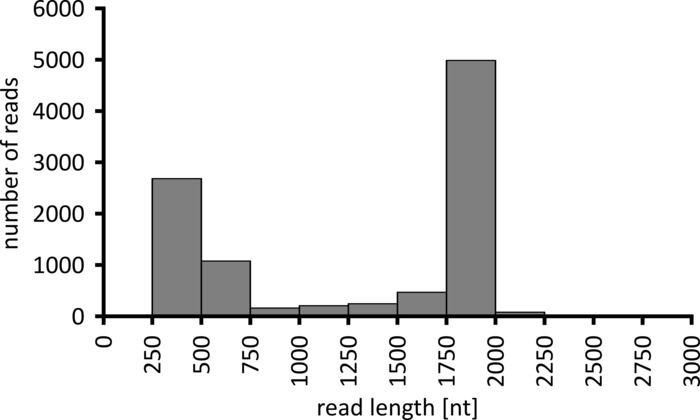
Figure 3: Read-length distribution of 10,000 reads from the representative experiment. The number of reads obtained having a given read length interval is indicated. Please click here to view a larger version of this figure.
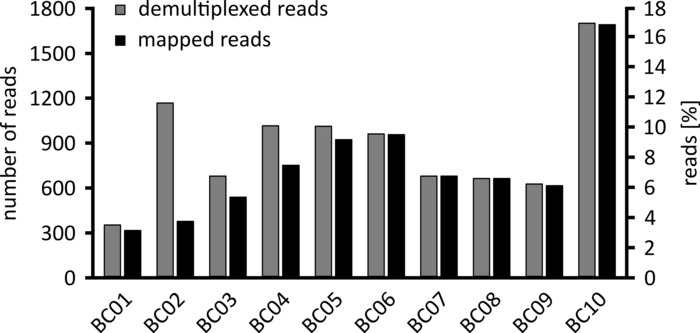
Figure 4: Distribution of reads after demultiplexing. The number and percentage of demultiplexed (grey) and mapped reads (black) for each barcode are shown. Please click here to view a larger version of this figure.
Table 1: Overview of primer sets used. Initial amplification of target sequences was performed with Primer Set 1. Primer Set 2 was then used for nested amplification and adapter addition. Adapters are indicated in red. Please click here to download this file.
Table 2: Overview of barcode sequences. Individual barcodes were used to identify each sequenced sample. Please click here to download this file.
Table 3: RNA concentrations obtained following extraction from blood samples sequenced in the representative experiment. The RNA concentrations of two individuals from each of five species are shown, and the ratios of the optical densities at 260/280 nm and 260/230 nm are indicated. Please click here to download this file.
