A Guide to Build a Highly Inclined Swept Tile Microscope for Extended Field-of-view Single-molecule Imaging
Özet
A detailed instruction is described on how to build a highly inclined swept tile (HIST) microscope and its usage for single-molecule imaging.
Abstract
Single-molecule imaging has greatly advanced our understanding of molecular mechanisms in biological studies. However, it has been challenging to obtain large field-of-view, high-contrast images in thick cells and tissues. Here, we introduce highly inclined swept tile (HIST) microscopy that overcomes this problem. A pair of cylindrical lenses was implemented to generate an elongated excitation beam that was scanned over a large imaging area via a fast galvo mirror. A 4f configuration was used to position optical components. A scientific complementary metal-oxide semiconductor camera detected the fluorescence signal and blocked the out-of-focus background with a dynamic confocal slit synchronized with the beam sweeping. We present a step-by-step instruction on building the HIST microscope with all basic components.
Introduction
Single-molecule fluorescence imaging plays an important role in many biological studies that reveal ultrastructures, dynamics and the quantity of biomolecules1,2,3. However, it has been challenging to study single-molecules inside cells or tissues. While confocal microscopy provides high sectioning capability4, it is not suitable for single-molecule imaging due to severe photobleaching by the high excitation intensity or slow imaging speed. Widefield microscopy uses weaker illumination but suffers from a poor signal to background ratio (SBR)5. Light-sheet microscopy, on the other hand, could show good sectioning and low photobleaching6; however, the available numerical aperture (NA) is greatly limited by the requirement of orthogonally placed objectives7. Alternatively, it requires special illuminators and sample chambers8,9.
For these reasons, highly inclined and laminated optical sheet (HILO) microscopy has been widely used for 3D single-molecule imaging10. When an inclined beam encounters an interface of two media (glass and water, for example), the beam is refracted according to Snell's law. Importantly, the refracted beam gets thinner, and its thickness is described as dz = R/tan(θ) where R is the diameter of the inclined beam and θ is the refraction angle of the transmitted beam. This simple implementation results in a good sectioning capability. Nevertheless, this relation indicates that a thin illumination (i.e., high sectioning capability) requires a small R and/or a large θ. For example, when R = 20 µm and θ = 72 degrees, one can obtain dz = 6.5 µm. Since there is a practical limit to increasing the refraction angle in order to image deep inside cells and avoid total internal reflection, there is a strong coupling of the illumination diameter and the beam thickness. For this reason, HILO imaging shows a relatively small field-of-view (FOV) that greatly restricts its applications in multicellular imaging.
Recently, we have overcome this problem by highly inclined swept tile (HIST) microscopy where the FOV is decoupled from the beam thickness in a very simple way11. First, a beam elongated in one direction is generated via a pair of cylindrical lenses. This beam, termed as a tile, produces a thin illumination with dz ~ 4 µm while its FOV is 130 x 12 µm2. Then, the tile is swept across the sample using a rotating galvo mirror. Meanwhile, the fluorescence image is recorded on a scientific complementary metal-oxide semiconductor (sCMOS) camera that filters efficiently out-of-focus background by operating in a rolling shutter mode which serves as tunable confocal slit detection. In this way, HIST microscopy enables single-molecule imaging with a larger field of view (~130 x 130 µm2) and a thinner illumination than HILO imaging. We applied this new imaging technique to detect RNA transcripts with a single probe in cells or with a few probes in mouse brain tissues, which has significant potential for studying gene expression and diseases. Unlike other approaches, HIST employs only a single high numerical aperture objective without an additional illuminator or remote detection objectives and is fully compatible with inverted microscopes. These advantages along with a large FOV and high contrast will make HIST microscopy a prominent tool in biology and medicine. We present detailed instructions regarding instrumentation of the HIST microscope, and how to test and calibrate its performance as below.
Protocol
1. Setting up the microscope, lasers and alignment tools
- Before building the microscope, prepare all the necessary components including optics, optomechanics and electronics as listed in Table of Materials.
- Prepare a microscope body mainly composed of two parts: an objective holder with an RMS-threaded port and a piezo-stage mounted on an aluminum block (Figure 1A).
NOTE: The custom-made microscope body is used for the convenience and flexibility of instrumentation12. Any commercially available microscope body can be used for HIST microscopy. - Combining multiple laser lines and coupling them to a single mode fiber
- Install 405, 561, 638 nm lasers onto the optical table and combine the beams through a polarizing beam splitter and a long-pass dichroic mirror as shown in Figure 1B. Make sure that all laser beams pass through pinholes on the alignment tool. Insert half wave plates for power adjustment.
NOTE: Wear safety goggles for eye protection and use beam-blocks to absorb unwanted laser beams. - Install a fiber coupling lens (f = 4.5 mm) and a fiber adapter held in z-axis translator with a cage system.
- Connect a multimode fiber (MMF, Ø 62.5 µm) to the fiber adapter. Adjust each pair of the steering mirror and the z-translator until the coupling efficiency of each laser is higher than 95%. The output beam has a near Gaussian-shaped profile with speckle patterns.
- Take away the multimode fiber and connect a single mode fiber (SMF). Similar to MMF, fine-tune and maximize the coupling efficiency of three lasers.
- Install 405, 561, 638 nm lasers onto the optical table and combine the beams through a polarizing beam splitter and a long-pass dichroic mirror as shown in Figure 1B. Make sure that all laser beams pass through pinholes on the alignment tool. Insert half wave plates for power adjustment.
- Assemble a collimated light source that will be used for beam alignment in the excitation and detection paths. This device is composed of a temporally coherent light source (561 nm) connected to SMF, fiber adapter, achromatic lens (f = 60 mm), iris and Ø1" tube spacer in a cage system (Figure 1C). Adjust the distance between the fiber adapter and the lens using a shearing interferometer to ensure the collimation.
- Prepare a beam alignment tool (Figure 1D). This is a pair of aluminum posts with pinholes at 2" height from the surface of optical table, which allows for quick and precise beam alignment.
- Assemble a double pinhole system which consists of two Ø1" ground glass alignment disks at each end and two Ø1" lens tubes (the bottom one is slotted) as shown in Figure 1E.
2. Setting up the detection path
- Take out the objective and install the collimated light source. Adjust the knobs of the mirror (M1) beneath the objective holder so that the output beam from the microscope is roughly parallel to the optical table in height and aligned with threaded holes on the table. Refer to Figure 2 for detailed positions of each optical component.
- Insert a multiband dichroic mirror (DM) and reflect the beam by 90 degrees. Use the maximum size of the iris and make sure that the beam passes through the center of the dichroic mirror without clipping.
- Use the leaky beam passing through the dichroic mirror to guide the alignment of detection path. Place an sCMOS camera into the beam and make sure that the beam hits the center of the camera chip by using two mirrors (M2 and M3).
- Insert a tube lens (TL; f = 300 mm) approximately 300 mm away from the camera.
- Remove the collimated light source and adjust the relative distance between the tube lens and the camera until a pattern on the ceiling is clearly resolved by the camera.
- Insert a multi-band pass filter (BF) before the tube lens for multi-color fluorescence imaging.
3. Setting up the excitation path
- Reinstall the collimated light source on the objective holder. Place a fold-mirror (M4) to redirect the beam output from the microscope by 90 degrees. Adjust the knobs of the dichroic mirror and the fold-mirror iteratively until the beam passes through pinholes in the beam alignment tool.
- Detach the collimated light source and install it on the table, in which the beam points towards the microscope body. Align the beam using a beam alignment tool and a double pinhole system.
- Insert a lens L4 (f = 400 mm; Ø = 2") to the optical path approximately 400 mm away from the objective holder. Install the objective lens and adjust the position of L4 along the optical axis until a perfect Airy disk pattern is formed on the ceiling.
NOTE: When inserting the lens, the beam position passing through the lens should be kept unchanged. The lens L4 has a SM2 thread that allows it to be attached/detached from the 60 mm SM2-threaded cage plate easily. - Unscrew the objective and reinstall the collimated light source with an open iris. Trace down the beam output from the microscope with a business card. Mount a mirror M5 at the place where the size of beam is the smallest and approximately 400 mm away from L4, which is a conjugated image plane (cIP).
- Install a mirror M6 and reflect the beam by 90 degrees. Adjust M5 and M6 iteratively with the beam alignment tool.
- Insert a lens L3 (f = 150 mm) roughly 150 mm away from M5. Use a shearing interferometer to ensure the collimation of the output beam.
- Temporarily take away L4 and trace down the beam to find the focal position of L3. Put a single axis galvo mirror at this point, which is a conjugated back focal plane (cBFP). Supply 0 volts to the galvo mirror and rotate the holder of the galvo mirror so that it reflects the beam by 90 degrees.
- Place a fold-mirror M7. Properly place L2 (f = 100 mm) the same way as step 3.6.
- Remove the collimated light source from the object holder. Install a collimation lens L1 (f = 100 mm), fiber adapter and iris. Connect a single mode fiber to the adapter and send the beam through the imaging system.
- Reinsert L4 and fine tune the system until a perfect Airy disk pattern appears on the ceiling.
4. Setting up the cylindrical lenses
- Insert a cylindrical lens (CL1, f = 400 mm) after L1 and make sure that the cylindrical lens focuses the beam along the x-axis.
- Insert another cylindrical lens (CL2, f = 50 mm) to the beam path. Use a shearing interferometer to ensure the output beam is collimated.
NOTE: The distance between the two cylindrical lenses is 450 mm. The output beam has a compression ratio of 8 and an elongated oval shaped Airy disk pattern is formed on the ceiling.
5. Testing tile imaging
- Prepare 3D hydrogel sample. Mix 20 nm Crimson beads with a hydrogel solution, which consists of 12% acrylamide:bisacrylamide (29:1), 0.2% (v/v) TEMED and 0.2% (w/v) ammonium persulfate in 0.75x TAE buffer. Inject 50 µL of the mixed solution into a flow chamber as described elsewhere11. After 10 min, the 3D hydrogel sample is ready to for imaging.
- Put the sample onto the sample holder. Turn on 638 nm laser and adjust the power to <1 mW for sample excitation.
- Run the camera control software. In the camera acquisition-setting panel, select Internal in trigger mode and then click Take video for free running mode.
- Slightly adjust the position of the camera so that the image is located at the center of the camera when 0 volts is applied to the galvo mirror.
- Rotate the horizontal knob of the mirror M5 to achieve a highly inclined illumination.
NOTE: With the increase of the illumination angle, the hydrogel image becomes clearer than that of the Epi image as the beam gets thinner. However, the image keeps almost the same position. - Record the tile image. Calculate the effective illumination width11. For example, Figure 3 shows the effective illumination width of 12 µm.
6. HIST imaging
- Prepare a data acquisition board connected with a terminal block. Connect USER 1 BNC connector with P0.0 through an electrical wire. Use USER 1 as a digital output for the external triggering of sCMOS camera. Connect an analog output AO0 to a galvo mirror driver.
- Generate TTL pulse trains from P0.0 using a custom-made program (Figure 4A) and set the period time = 400 ms and t_ON = 2 ms. Check the generated pulses from USER 1 BNC terminal by a digital oscilloscope and then connect the BNC cable to the camera external trigger port.
NOTE: The control software used in this paper is available upon request. When imaging at different camera frame rates, the period time should be adjusted accordingly. - Start sweeping a galvo mirror via the custom-made program. Adjust Vmin to -500 mV and Vmax to 500 mV for full FOV imaging. Note that under this operation, 3D hydrogel samples still show high background similar to Epi illumination.
- Changing the camera acquisition setting.
- Select External in trigger mode and Down (Sequential) in LightScan Plus drop-down menu as shown in Figure 4B.
NOTE: In this setting, the camera does not take images unless a trigger signal is turned on. - Click Scan Speed Control for window height and line exposure time control and set the values to be 180 rows and 28 ms, respectively.
NOTE: When a tile width (Weff) is 180 rows (12 µm) and an integration time per line (Tint) is 28 ms, a delay time between lines (TD) is determined as TD = Tint/Weff = 0.156 ms. For imaging 2,048 x 2,048 pixels, the total acquisition time is 2,048 x TD + Tint = 346 ms, corresponding to ~2.9 fps.
- Select External in trigger mode and Down (Sequential) in LightScan Plus drop-down menu as shown in Figure 4B.
- Slightly adjust Vmax and Vmin to obtain clearer images.
- Obtain 3D stack images using the custom-made program by switching on 3D stack ON and specifying the number of stacks and the step size.
Representative Results
As an example, single-stranded DNA labeled with Atto647N was imaged with an excitation wavelength of 638 nm in a 3D hydrogel. DNA was anchored to the hydrogel network via an acrydite moiety during gel polymerization. The images were taken at 5 µm above the surface as shown in Figure 5a. The HIST image showed much less background compared to the Epi image, from which the signal to background ratio was calculated to be 1.9 ± 0.7 for the HIST image while most of the single molecule spots could barely be detected by Epi.
Single-molecule RNA fluorescence in situ hybridization (smFISH) was performed with 4 FISH probes. Figure 5b displays smFISH images of EEF2 (eukaryotic translation elongation factor 2) labeled with AlexaFluor 647 on A549 cells in an imaging buffer (refer to our previous work regarding the sample preparation11). A maximum intensity projection was performed on 20 z-stacks corresponding to 5 µm thickness. The HIST image showed not only much improved SBR but also more uniform illumination compared to Epi image. For Epi imaging, the exposure time was 400 ms while for HIST imaging the integration time per line was 32 ms, both of which had the same illumination power of 7.5 mW measured before the objective. The imaging speeds of Epi and HIST were 2.5 fps.

Figure 1. Microscope body, lasers and alignment tools. (A) Objective and sample holder. (B) Photo of laser systems. LP, long-pass dichroic mirror; λ/2, half-wave plates; PBS, polarizing beamsplitter. (C) Collimated light source. (D) Beam alignment tool with two insertable pinholes. (E) Double pinhole system. Please click here to view a larger version of this figure.
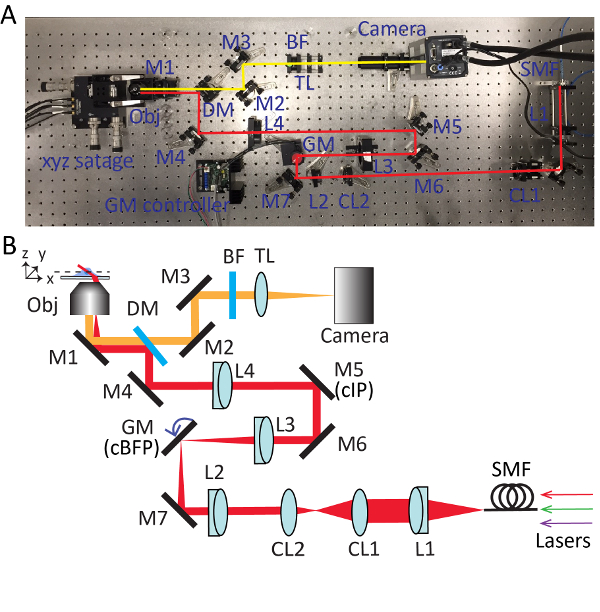
Figure 2. Detailed setup for highly inclined swept tile (HIST) microscopy. Photo (A) and schematic (B) of HIST microscope system. BF, multi-band pass filter; CL1-2, cylindrical lenses; DM, dichroic mirror; GM, galvo mirror; BF, band-pass filter; M1-7, mirrors; L1-4, lenses; SMF, single mode fiber; TL, tube lens; cIP, conjugated image plane; cBFP, conjugated back focal plane. Please click here to view a larger version of this figure.
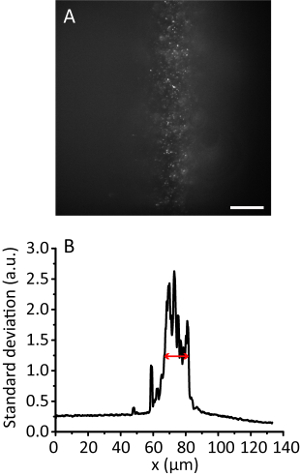
Figure 3. Tile illumination with a compression ratio of 8. (A) Fluorescence image of 20 nm beads in a 3D hydrogel. Scale bar, 20 µm. (B) Standard deviation projection along the y direction of A, smoothed by 10 data points. The red arrow indicates an effective illumination width of 12 µm. Please click here to view a larger version of this figure.
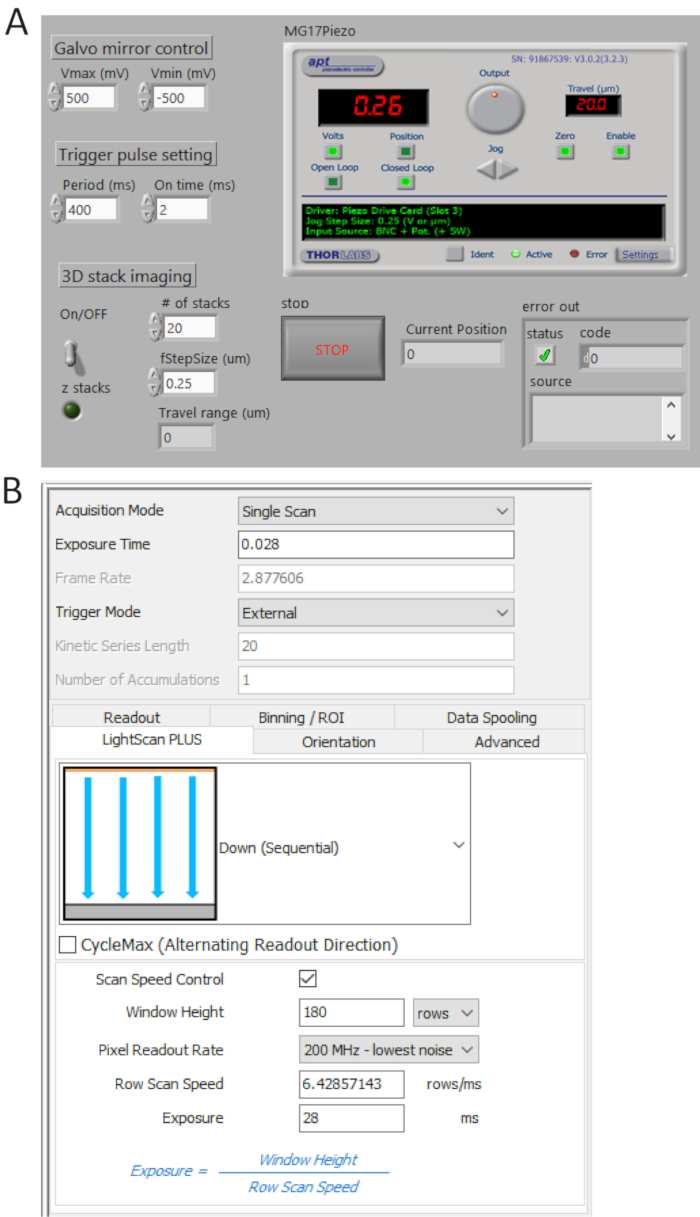
Figure 4. Control and imaging software front panels. (A) A custom-made LabView program synchronously controls the scanning of the galvo mirror, the starting acquisition of sCMOS camera and the movement of the piezo stage. (B) Camera acquisition setting control panel. Please click here to view a larger version of this figure.
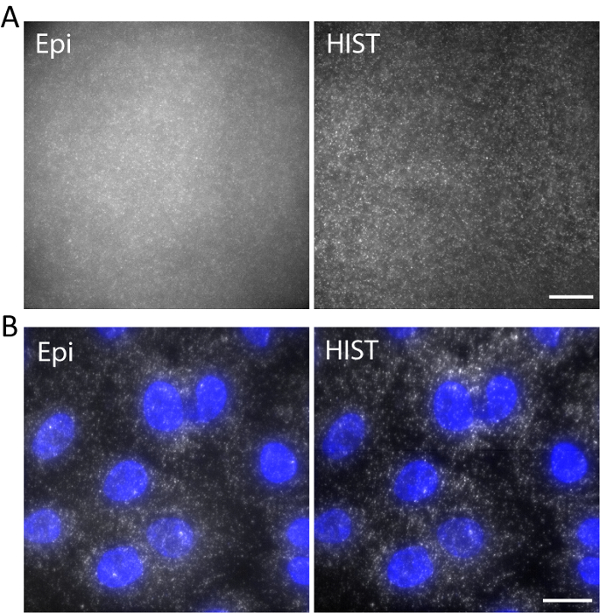
Figure 5. (A) Images of Atto647N-labeled DNA in a 3D hydrogel with Epi and HIST illumination. (B) smFISH images of EEF2 using 4 FISH probes on A549 cells by Epi and HIST microscopy. DAPI stain is shown in blue. Scale bars, 20 µm. Please click here to view a larger version of this figure.
Discussion
There are two critical steps in this protocol. The first one is the proper placement of L4 in step 3.3, ensuring that the incident beam passes through the center of the lens and a perfect Airy disk pattern is formed on the ceiling. The position of L4 determines the placement of all the other optical components, including M5, L3, GM and L2. The second critical step is the synchronization process. To reject the out of focus background, active pixels whose effective detection width is equal to the tile width should be synchronized with the beam sweeping. Therefore, it is necessary to measure the effective illumination width of a tile beam (step 5.6) and set camera parameters accordingly in step 6.4.
When imaging with very large FOV, the presented method shows an increased background at one side compared to the other side. This is attributed to slightly altered angles of illumination at different imaging positions. Implementing a second galvo mirror instead of M5 alleviates this problem as demonstrated before by synchronously adjusting the position and the scanning angle11. Instead of off-the-shelf achromatic doublets, a telecentric scan lens will be also helpful. However, for imaging an area of <8,080 µm2, single galvo mirror sweeping was sufficient. HIST microscopy has a limit of the imaging depth, however, it is able to obtain a good SBR when imaging up to ~15 µm with a 12 µm tile beam and a NA 1.45 oil immersion objective lens11.
In this protocol, we used a beam compression ratio of 8 to make a tile beam. A thinner illumination can be used in HIST microscopy to achieve higher SBR, which might be powerful for single-molecule tissue imaging11. However, in this case, photobleaching effect should be considered by an increased excitation intensity while the current beam compression ratio showed reduced photobleaching in 3D imaging compared to Epi11. Compared to light-sheet microscopes with two orthogonally placed objectives, HIST microscopy is simple to implement and compatible with conventional sample preparations. The enhanced SBR and large FOV of HIST microscopy is suitable for studying the interactions and dynamics of single biomolecules in multiple cells and can be used further in super-resolution imaging and single-molecule tracking.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
This work was supported by Defense Advanced Research Projects Agency (DARPA) (HR00111720066) and National Science Foundation (NSF) (1805200). We thank Michael Serge in Andor Technology for generously loaning the sCMOS camera.
Materials
| 1" Achromatic doublet | Thorlabs | AC254-060-A-ML | Collimator |
| 1" Achromatic doublet | Thorlabs | AC254-100-A-ML | L1,L2 |
| 1" Achromatic doublet | Thorlabs | AC254-300-A-ML | TL |
| 1" Broadband Dielectric Mirrors | Thorlabs | BB1-E02-10 | M1~M7 |
| 1" Cylindrical Lenses | Thorlabs | LJ1363RM-A | CL1 |
| 1" Cylindrical Lenses | Thorlabs | LJ1695RM-A | CL2 |
| 1" square kinematic mount | Edmund Optics | 58-857 | For dichroic mirror mounting |
| 1" Threaded Cage Plate | Thorlabs | CP02 | For holding other lenses |
| 2" Achromatic doublet | Thorlabs | AC508-150-A-ML | L3 |
| 2" Achromatic doublet | Thorlabs | AC508-400-A-ML | L4 |
| 2" Threaded Cage Plate | Thorlabs | LCP01 | For holding L4 |
| 2" Threaded Cage Plate | Thorlabs | LCP01T | For holding L3 |
| 2% Bis Solution | Bio Rad | 64085292 | hydrogel component |
| 20 nm fluorescent beads | Thermo Fisher | F8782 | For testing imaging |
| 30 mm Cage Right-Angle Kinematic Mirror Mount | Thorlabs | KCB1 | For objective & camera mounting |
| 30mm Cage System Iris | Thorlabs | CP20S | |
| 3-Axis NanoMax Stage | Thorlabs | MAX311D | |
| 40% Acrylamide Solution | Bio Rad | 64148001 | hydrogel component |
| 405 nm laser | Cobolt | Cobolt 06-MLD | |
| 50x TAE buffer | Bio-Rad | 161-0743 | hydrogel component |
| 561 nm laser | Cobolt | Cobolt 06-DPL | |
| 638 nm laser | Cobolt | Cobolt 06-MLD | |
| Ammonium persulfate | Sigma | A3678-25G | hydrogel component |
| Beam alignment tool | custom made | ||
| BNC terminal blocks | Natural Instruments | BNC-2110 | |
| Cage plate with M9 x 0.5 internal threads | Thorlabs | CP1TM09 | For holding aspheric lens |
| Cage System Rods | Thorlabs | SR series | |
| Cell culture & smFISH | See a reference [11] | ||
| Double side tape | Scotch | 515182 | Flow chamber |
| Epoxy | Devcon | 14250 | Flow chamber |
| Galvo mirror | Thorlabs | GVS211 | GM |
| Galvo System Linear Power Supply | Thorlabs | GPS011 | |
| Half wave plate | Thorlabs | WPH10M-405/561/633 | Power adjustment |
| long-pass dichroic mirror | Chroma | T550lpxr | For combining lasers |
| Microscope slides | Fisherbrand | 12549-3 | Flow chamber |
| Mikroskopische Deckglaser | Hecht Assistent | 990/5024 | Flow chamber |
| Mounted Frosted Glass Alignment Disk | Thorlabs | DG10-1500-H1-MD | For double pinhole system |
| Mounted rochester aspheric lens | Thorlabs | A230TM-A | |
| Multi-band dichroic mirror | Semrock | Di03-R405/488/561/635-t3 | DM; 3 mm thickness |
| Multi-band filter | Semrock | FF01-446/523/600/677-25 | BF |
| Multimode fiber | Thorlabs | M31L02 | MMF |
| N,N,N',N'-tetramethyl ethylenediamine | Sigma | T7024-25ML | hydrogel component |
| NI-DAQ board | Natural Instruments | PCI-6733 | |
| Ø1" Kinematic Mirror Mount | Thorlabs | KM100 | For holding mirrors |
| Objective lens | Olympus | PLANAPO N 60X | 60X 1.45NA oil |
| Pedestal Base Clamping Forks | Newport | 9916 | |
| Pedestal Pillar Posts | Thorlabs | RS1P8E | |
| Piezo controller | Thorlabs | BPC303 | |
| Polarized beam splitter | Thorlabs | PBS251 | For combining lasers |
| RMS-SM1 adapter | Thorlabs | SM1A3TS | For objective lens |
| Rod holder | custom made | ||
| Rotation cage mount | Thorlabs | RSP1/CRM1/CRM1P | For HWP & cylindrical lens mounting |
| sCMOS camera | Andor | Zyla-4.2P-CL10 | |
| Shearing interferometer | Thorlabs | SI100 | Beam collimation test |
| Single mode fiber | Thorlabs | P5-405BPM-FC-2 | SMF |
| SM1 Lens Tubes | Thorlabs | SM1S25 | For double pinhole system |
| SM1 Slotted Lens Tube | Thorlabs | SM1L30C | For double pinhole system |
| Stage mount | custom made | ||
| threaded fiber adapter | Thorlabs | SM1FC | |
| Z-Axis Translation Mount | Thorlabs | SM1Z | Fiber coupling |
Referanslar
- Sigal, Y. M., Zhou, R., Zhuang, X. Visualizing and discovering cellular structures with super-resolution microscopy. Science. 361, 880-887 (2018).
- Lerner, E., et al. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science. 359, (2018).
- Raj, A., van Oudenaarden, A. Single-Molecule Approaches to Stochastic Gene Expression. Annual Review of Biophysics. 38, 255-270 (2009).
- Wilson, T. Resolution and optical sectioning in the confocal microscope. Journal of microscopy. 244, 113-121 (2011).
- Sase, I., Miyata, H., Corrie, J. E., Craik, J. S., Kinosita, K. Real time imaging of single fluorophores on moving actin with an epifluorescence microscope. Biophysical Journal. 69, 323-328 (1995).
- Huisken, J., Swoger, J., Del Bene, F., Wittbrodt, J., Stelzer, E. H. K. Optical Sectioning Deep Inside Live Embryos by Selective Plane Illumination Microscopy. Science. 305, 1007-1009 (2004).
- Cella Zanacchi, F., et al. Live-cell 3D super-resolution imaging in thick biological samples. Nature Methods. 8, 1047 (2011).
- Galland, R., et al. 3D high- and super-resolution imaging using single-objective SPIM. Nature Methods. 12, 641 (2015).
- Gustavsson, A. K., Petrov, P. N., Lee, M. Y., Shechtman, Y., Moerner, W. E. 3D single-molecule super-resolution microscopy with a tilted light sheet. Nature Communications. 9, 123 (2018).
- Tokunaga, M., Imamoto, N., Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature Methods. 5, 159-161 (2008).
- Tang, J., Han, K. Y. Extended field-of-view single-molecule imaging by highly inclined swept illumination. Optica. 5, 1063-1069 (2018).
- Han, K. Y., Kim, S. K., Eggeling, C., Hell, S. W. Metastable Dark States Enable Ground State Depletion Microscopy of Nitrogen Vacancy Centers in Diamond with Diffraction-Unlimited Resolution. Nano Letters. 10, 3199-3203 (2010).
- Sinkó, J., Szabó, G., Erdélyi, M. Ray tracing analysis of inclined illumination techniques. Optics Express. 22, 18940-18948 (2014).
