The protocol outlined in Figure 1 describes the basic workflow for running CETSA assays on adherent cells with detection of remaining soluble protein by high content imaging. This workflow can be easily adapted to all stages of assay development by modifying the plate layout of the compounds or reagents14. We detail expected results for several anticipated use cases below.
Antibody identification and assay development. A prerequisite for successful results is the identification of a primary antibody or other suitable affinity reagent that selectively recognizes the native form of the protein in the presence of the aggregated and precipitated protein formed during the heat challenge in step 3. To establish the CETSA imaging assay described here, we screened a panel of 9 antibodies targeting p38α at 52°C for signal window between the positive and negative controls. We then titrated the best antibodies and settled on the conditions shown in Figure 2 A,B with representative immunofluorescence images for p38α stabilized by a known ligand (positive control) and DMSO (negative control). Antibody recognition should also not be disrupted by the conformational changes of the target protein that may be induced by ligand binding (Figure 2C). As an example, BIRB796 has a long off rate, and quantification of target engagement was only possible by applying an antigen retrieval step (5.2; Figure 2D). It is important to validate the performance of the primary antibody with known ligands covering different binding sites of the target protein if available. The antibody validation is preferably done both with and without the antigen retrieval step.
Tagg and ITDRF curves. As mentioned above, CETSA experiments can be run in two different modes, Tagg curves and ITDRF experiments. Both variants utilize the same basic protocol outlined in Figure 1 and in the protocol section. In the first setup, the purpose is to challenge the cells with a temperature gradient and compare the Tagg curves in the presence and absence of a single concentration of ligand. To perform a Tagg curve, separate plates are heated for 3 minutes across a range of temperatures. In performing this experiment, it is important to time the compound treatment length for each plate with the time it takes for the water bath to stabilize to the new temperature. In this regard, peforming the heat challenge step in the water bath is more time consuming than heating tubes in a PCR machine. The experimental path is to next run concentration response curves of a ligand at a fixed temperature to generate ITDRF curves. In general, when testing multiple compounds, assay ready plates for the compound addition are prepared using automated liquid handling to achieve the most reproducible data. Compounds are serially diluted in DMSO and then dissolved in cell culture media to the desired concentrations. We have typically tested 11 point concentration series starting at 50 – 100 µM in 3 or 4 fold dilution, but this depends on the potency of the ligands used. It is advised to first establish the Tagg curve both in absence and presence of a ligand and select the temperature for subsequent isothermal experiments where a shift between the curves can be observed. The selected temperature should be around or just above the Tagg. Both formats allow for confirmation of target engagement but for ranking of compound affinities ITDRF experiments are often more suitable. Figure 3A shows an illustration of anticipated quantified results for a Tagg curve and Figure 3B quantifies results of a typical ITDRF experiment.
Screening campaign. The protocol can also be adapted to screening campaigns to identify novel binders of the target protein. In this case, isothermal heat challenges are applied for a large number of compounds at a single concentration followed by ITDRF experiments for identified stabilizing compounds. We prepare assay ready screening plates and transfer the compounds diluted in culture media to the assay plates using automated liquid handling. As with all thermal stability assays, it is necessary to exceed the dissociation constant to observe protein stabilization, and thus we have applied small molecule libraries at 50 µM to facilitate hit identification. Triaging of the hits using ITDRF will later allow ranking and prioritization of these compounds. Figure 4 shows a representative result from a screening plate.
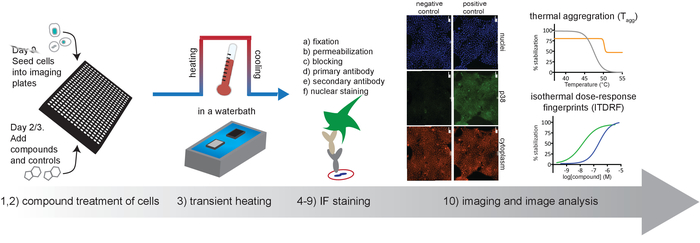
Figure 1. Schematic overview of the protocol described in this article. Please click here to view a larger version of this figure.
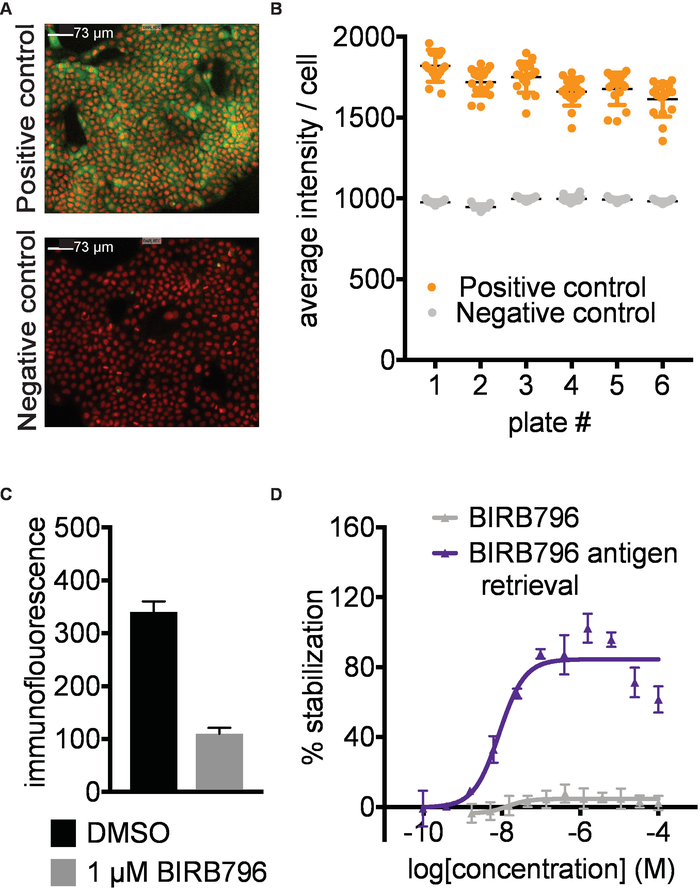
Figure 2. Antibody identification and assay development. A) Example data for detection of human p38α in A-431 cells. Representative images of positive (1 µM AMG548) and negative (DMSO) controls. Red- nuclear Hoechst staining, Green- p38α staining. Images taken with 10X magnification; the white scale bar represents 73 µm. B) Example of quantified data expressed as average intensity/cell. Error bars represent standard error of mean of 16 replicates for 6 separate plates. Each plate was heated to 52 °C in a water bath followed by fixation of the cells, permeabilization and subsequent immunostaining. C) Immunofluorescence signal intensity measured after treating A-431 cells with 1 µM BIRB796 or DMSO as described above followed directly by fixation. In the absence of a heating step, the BIRB796 signal is lower than the DMSO signal, suggesting that BIRB796 disrupts the detection of p38α with this antibody. D) ITDRFCETSA curves for cells treated with BIRB796 either with (blue triangle) or without (grey triangle) antigen retrieval protocol. This figure has been modified from Axelsson et al. 201812. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
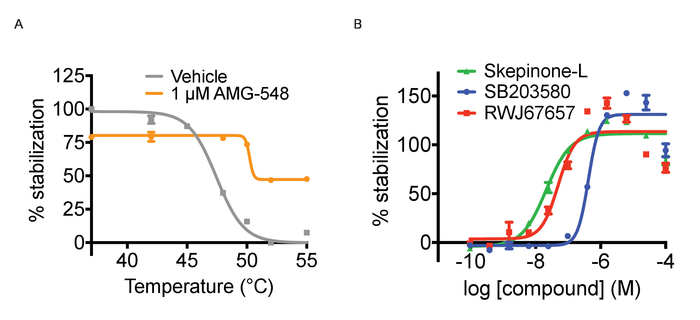
Figure 3. Thermal aggregation and ITDRF experiments. A) Thermal aggregation curve experiments for cells treated with positive (1 µM AMG548, in orange) and negative (DMSO in grey) controls. Error bars represent standard error of mean of 32 or 464 replicates. B) ITDRFCETSA experiment performed at 52 °C for cells treated with serial dilutions of SB203580 in blue, Skepinone-L in green and RWJ67657 in red. Error bars represent standard error of the mean of 6 replicates. Quantified data are expressed as average intensity/cell and normalized against the highest concentration of respective compound. This figure has been modified from Axelsson et al. 201812. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
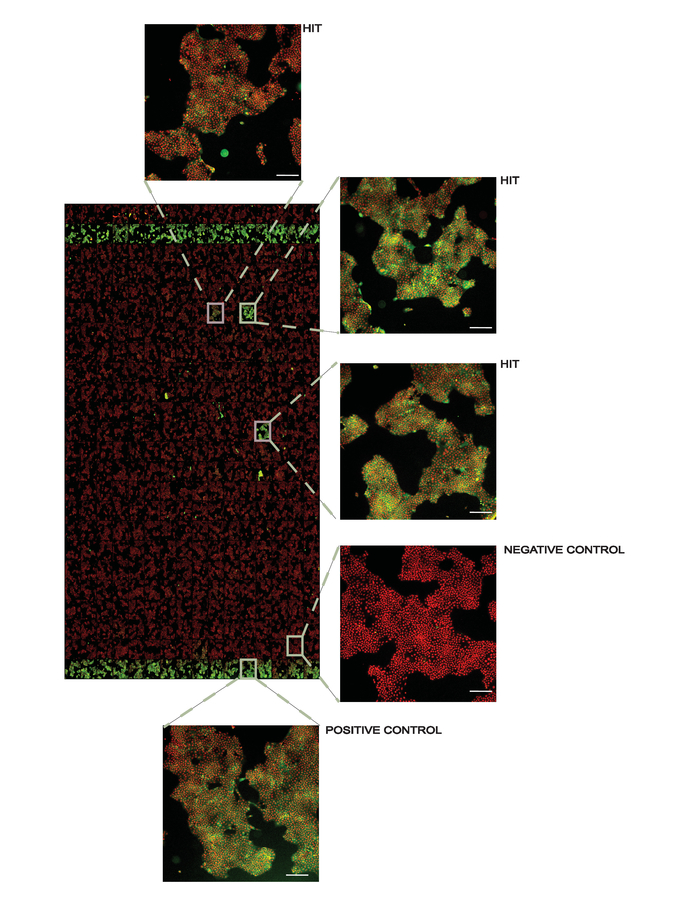
Figure 4. Representative overview of a screening plate at 10X magnification. Positive controls (1 µM AMG548) are in columns 2 and 24 and negative controls (DMSO) are in columns 1 and 23. All other wells contain 50 µM of the library compounds used for screening with several hits denoted. In the inset figures, the white line in the lower right corner represents 200 µm. This figure has been modified from Axelsson et al. 201812. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.
