“Liver-on-a-Chip” Cultures of Primary Hepatocytes and Kupffer Cells for Hepatitis B Virus Infection
Özet
The goal of this protocol is to provide a step-by-step guide to perform 3-D "liver-on-a-chip" infection experiments with the hepatitis B virus.
Abstract
Despite the exceptional infectivity of the hepatitis B virus (HBV) in vivo, where only three viral genomes can result in a chronicity of experimentally infected chimpanzees, most in vitro models require several hundreds to thousands of viral genomes per cell in order to initiate a transient infection. Additionally, static 2D cultures of primary human hepatocytes (PHH) allow only short-term studies due to their rapid dedifferentiation. Here, we describe 3D liver-on-a-chip cultures of PHH, either in monocultures or in cocultures with other nonparenchymal liver-resident cells. These offer a significant improvement to studying long-term HBV infections with physiological host cell responses. In addition to facilitating drug efficacy studies, toxicological analysis, and investigations into pathogenesis, these microfluidic culture systems enable the evaluation of curative therapies for HBV infection aimed at eliminating covalently closed, circular (ccc)DNA. This presented method describes the set-up of PHH monocultures and PHH/Kupffer cell co-cultures, their infection with purified HBV, and the analysis of host responses. This method is particularly applicable to the evaluation of long-term effects of HBV infection, treatment combinations, and pathogenesis.
Introduction
The study of HBV has been complicated by the poor susceptibility of culture systems, requiring several hundreds to thousands of HBV genome copies per cell to initiate the infection1. Furthermore, primary human hepatocytes are generally exceptionally fragile and rapidly dedifferentiate during conventional cultures2. This is mainly due to the fact that the flat and hard plastic surfaces do not mimic the natural extracellular environments found within the liver and the general lack of oxygenation of the cultures in the absence of microfluidic circulation. Conventional static hepatocyte cultures on collagen-coated plates rapidly dedifferentiate and lose their susceptibility to HBV infection3. Here, we describe the set-up and infection of PHH grown in 3D liver-on-a-chip cultures, which are vastly advantageous over conventional 2D static PHH cultures on collagen-coated plates due to their extended metabolic and functional competence, facilitating long-term cultures of at least 40 days4. In this system, PHH are seeded on collagen-coated scaffolds, which are continually perfused with growth medium to supply oxygen and nutrients to the cells. Even though alternative culture systems for PHH based on complex cocultures of murine fibroblasts or 3D growth in spheroids have been validated and are susceptible to HBV infection using multiplicities of infection of 500 genome equivalents (GE) of HBV per cell, 3D liver-on-a-chip cultures remain the sole in vitro model system susceptible to 0.05 GE of HBV per cell4. This is additionally underpinned by the necessity of using high concentrations of dimethyl sulfoxide (DMSO) and polyethylene glycol (PEG) to establish HBV infection in these cultures, which is dispensable for the infection of 3D liver-on-a-chip culture systems4. Among the major hallmarks of HBV infection is the cccDNA pool, which acts as the transcriptional template for all de novo–produced virions5,6. Even though cccDNA can be detected in conventional hepatocyte cultures7,8, it remains unclear as to whether the regulation of cccDNA and any therapeutic approaches aimed at its elimination are recapitulated in partially or completely dedifferentiated hepatocytes. We have shown that cccDNA is functionally formed in 3D liver-on-a-chip cultures, responds to physiological stimuli, and can be targeted by interfering with the accessibility of the transcriptional machinery to the cccDNA genome4.
Host responses to HBV infection in 3D liver-on-a-chip mimic those observed in HBV-infected patients, enabling the identification of biomarkers for infection, as well as therapeutic success. Among the unique features of liver-on-a-chip cultures is the ability to evaluate long-term host responses between PHH and other nonparenchymal cells within the liver, including Kupffer cells4, stellate cells9, and liver sinusoidal endothelial cells10,11. This offers the unique opportunity to evaluate cell/cell interactions in a complex 3D microenvironment.
Additionally, the extended culture period of this platform facilitates the evaluation of sequential drug treatments and their impact on HBV persistence, which are not possible using conventional hepatocyte culture systems.
This protocol describes how 3D liver-on-a-chip cultures are generated, either for monocultures of PHH or for cocultures of PHH with Kupffer cells. Furthermore, we describe the production of purified HBV for low-multiplicity-of-infection studies, as well as the subsequent analysis of host and viral responses.
Protocol
1. Assembly and Equilibration of Plates
- Ensure that both the compressor and the vacuum pump associated with the LiverChip platform are turned on. Perform the assembly and equilibration of the plates in a class II cabinet.
- Aseptically assemble the microfluidic plates by placing a sterile membrane between the plate base and adding the well-containing top plate (Figure 1a).
- Ensure that the sterile membrane smoothly rests on the two pins of the base plate, since uneven membrane placement compromises the microfluidic circulation.
- Add a sterile plate lid and tighten the screws at the base of the plate using an automated precision torque to 33 lb using a spiral tightening sequence. Ensure that all screws are tightened to 35 lb using a manual torque. During this step, ensure the screws are tightened symmetrically (Figure 1a).
- Prewarm hepatocyte seeding medium containing Williams E medium, primary hepatocyte thawing, and plating supplements, 5% fetal bovine serum (FBS), and 1 µM dexamethasone to 37 °C before priming.
- Prime the completely assembled plate by placing it within the washing dock and adding 400 µL of hepatocyte seeding medium to the reservoir side of each well. Ensure the plate snaps into the washing dock completely.
- Initiate flow in the upward direction for 3.5 min at 1 µL/s. The successful function of the microfluidic circulation can be ascertained through the red indicators at the side of the plate (Figure 1a).
- Once the medium is pumped to the cell growth side of the plate, ensuring the correct assembly of the flow channel, add an additional 1.2 mL of hepatocyte seeding medium.
- Carefully transfer the plate into the docking station within a humidified incubator at 37 °C and 5% CO2 and initiate flow in the upward direction at a flow rate of 1 µL/s for 16 h (Figure 1c).
- Transfer the plate to the washing dock and eliminate bubbles in the well by gently pipetting up and down.
- Add one sterile, round filter paper, followed by a cell attachment scaffold and a retaining ring, to each well using sterile forceps. Press down each well with a sterile plunger to lock the retaining rings and scaffolds into place (Figure 1b).
- Aspirate all medium and add 400 µL of prewarmed hepatocyte seeding medium gently over the scaffold and initiate flow in the downward direction for 3.5 min at 1 µL/s.
- Aspirate all medium pumped out of the reservoir side of the plate. This step is necessary to replace the medium contained in the flow channel.
- Add 1.4 mL of hepatocyte seeding medium to each well before returning the plate to the dock to complete the total volume per well. The volume per well is now 1.6 mL (1.4 mL in the well and 0.2 mL in the flow channel).
2. Thawing and Seeding of Hepatocytes for Monocultures
- Prewarm hepatocyte thawing medium and hepatocyte seeding medium to 37 °C prior to thawing one vial of PHH according to the suppliers' instructions. Use a centrifuge at room temperature during this step to avoid abrupt temperature changes. Perform the thawing and seeding of the hepatocytes in a class II cabinet.
- Resuspend the cells in 1 mL of hepatocyte seeding medium and count the cells using trypan blue. Ensure that the viability of the cells is above 90%.
- Keep the cells on ice until they are added to the wells.
- Transfer the equilibrated and fully assembled plate to the washing dock and aspirate all medium from the wells.
- Add 600,000 hepatocytes to each well in a 500 µL volume of hepatocyte seeding medium.
- Initiate flow in the downward direction at a flow rate of 1 µL/s and bring the total volume in the well to 1.6 mL by adding 900 µL of hepatocyte seeding medium.
- Transfer the plate to the docking station within a humidified incubator at 37 °C and with 5% CO2.
- Initiate flow in the downward direction at a flow rate of 1 µL/s for 8 h, followed by a flow reversal to the upward direction at a flow rate of 1 µL/s for 8 h.
- Transfer the plate to the washing dock and aspirate all medium from the wells.
- Add 400 µL of hepatocyte maintenance medium (Williams E medium supplemented with hepatocyte maintenance supplements and 100 nM dexamethasone) to each well and initiate flow in the downward direction at a flow rate of 1 µL/s for 3.5 min.
- Aspirate all medium from the reservoir and add 1.4 mL of hepatocyte maintenance medium (Figure 1d).
- Replace the medium with hepatocyte maintenance medium every 48 h. To ensure all medium in the well is replaced, perform a washing step before the addition of fresh hepatocyte maintenance medium.
- For the washing step, transfer the plate to the washing dock, aspirate all medium from the wells, and add 400 µL of maintenance medium.
- Initiate flow in the downward direction at 1 µL/s for 3.5 min. Aspirate all medium appearing on the reservoir side of the wells.
- Add 1.4 mL of hepatocyte maintenance medium and, within a humidified incubator at 37 °C and with 5% CO2, transfer the plate into the docking station and initiate flow in the upward direction at a flow rate of 1 µL/s for 48 h (Figure 1f).
NOTE: For hepatocyte monocultures used as controls for cocultures, in order to ensure controlled conditions, a second type of maintenance medium is used, which is specific for use in the cocultures with primary human Kupffer cells. 48 h after replacing the hepatocyte seeding medium with hepatocyte maintenance medium (day 3 post-seeding), the regular hepatocyte maintenance medium will be replaced with coculture maintenance medium II, especially in monocultures of PHH when comparing to cocultures of both PHH and Kupffer cells, as the medium components differ slightly.
3. Thawing and Seeding of Kupffer Cells and Hepatocytes for Co-cultures
- In order to ensure an accurate comparison of results, always compare PHH/Kupffer cell cocultures to PHH monocultures
- For cocultures of PHH and Kupffer cells, thaw one vial of Kupffer cells in advanced Dulbecco's modified Eagle's medium (AdDMEM) without dexamethasone but supplemented with primary hepatocyte thawing and plating supplements (coculture seeding medium) according to the suppliers' instructions. Perform the thawing and seeding of the Kupffer cells and hepatocytes in a class II cabinet.
- Resuspend the cells in 1 mL of coculture seeding medium and count the cells using trypan blue. Ensure that the viability of the cells is above 90%.
- Keep the cells on ice prior to adding them to the wells to avoid cell adhesion.
- Follow the instructions in steps 2.1–2.3 for the thawing of primary human hepatocytes.
- Transfer the equilibrated and fully assembled plate to the washing dock and aspirate all medium from the wells.
- Add 60,000 Kupffer cells and/or 600,000 hepatocytes to each well in a total volume of 250 µL each of coculture seeding medium.
- Initiate flow in the downward direction at a flow rate of 1 µL/s and add 900 µL of coculture seeding medium to each well.
- Transfer the plate into the docking station within a humidified incubator at 37 °C and with 5% CO2.
- Initiate flow in the downward direction at a flow rate of 1 µL/s for 8 h, followed by flow reversal to the upward direction at a flow rate of 1 µL/s for 8 h.
- Transfer the plate to the washing dock and aspirate all medium from the wells.
- Add 400 µL of coculture maintenance medium I (AdDMEM without dexamethasone but supplemented with hepatocyte maintenance supplements) to each well and initiate flow in the downward direction at a flow rate of 1 µL/s for 3.5 min.
- Aspirate all medium from the reservoir side and add 1.4 mL of coculture maintenance medium I to each well.
- Transfer the plate into the docking station within a humidified incubator at 37 °C and with 5% CO2 and initiate flow in the upward direction at a flow rate of 1 µL/s for 48 h.
- Transfer the plate to the washing dock and aspirate all medium from the wells. Add 400 µL of coculture maintenance medium II (Williams E medium without dexamethasone but supplemented with 100 nM hydrocortisone and hepatocyte maintenance supplements) and initiate flow in the downward direction at 1 µL/s for 3.5 min.
- Aspirate all medium appearing on the reservoir side of the wells.
- Add 1.4 mL of coculture maintenance medium II and transfer the plate into the docking station within a humidified incubator at 37 °C and with 5% CO2.
- Initiate flow in the upward direction at a flow rate of 1 µL/s for 48 h (Figure 1e).
- Replace the medium every 48 h with coculture maintenance medium II. To ensure all medium in the well is replaced, perform a washing step before the addition of fresh medium.
- To wash, transfer the plate to the washing dock, aspirate all medium from the wells, and add 400 µL of coculture maintenance medium II.
- Initiate flow in the downward direction at 1 µL/s for 3.5 min. Aspirate all medium appearing on the reservoir side of the wells.
- Add 1.4 mL of coculture maintenance medium II and transfer the plate into the docking station within a humidified incubator at 37 °C and with 5% CO2, and initiate flow in the upward direction at a flow rate of 1 µL/s for 48 h (Figure 1f).
4. Production of an Infectious Hepatitis B Virus for Infection Studies
- Perform this section of the protocol in a containment level III lab. Do the seeding, medium changes, medium collection, and virus concentration in a class II cabinet.
- Culture HBV-producing cells (e.g., HepDE19, HepAD38) in collagen-coated T1000 5-layer flasks in 120 mL of complete DMEM/F12 (10% FBS, penicillin/steptomycin, nonessential amino acids, 500 μg/mL G418, and 1 μg/mL tetracycline) until they reach 90% confluency.
- Change the medium to induction medium (complete DMEM without tetracycline) to induce the HBV production.
- Collect the complete medium volume every 48 h for 12 days post-withdrawal of tetracycline and store it at 4 °C.
- Filter the collected medium through a 0.45 µm bottle top filter.
- Add sterile PEG 8000 in phosphate-buffered saline (PBS) to the collected medium to a final concentration of 4% w/w, mix by inverting 8x–10x, and incubate at 4 °C for 16 h. Centrifuge at 10,000 x g for 1 h at 4 °C to collect the PEG-precipitated virus and resuspend the pellet in PBS containing 10% FBS.
- Combine the PEG-precipitated virus from all harvesting time points and layer it on top of a 20% sucrose cushion. Centrifuge at 140,000 x g for 16 h at 4 °C using an SW28 rotor.
- Aspirate the supernatant and resuspend the pellet in PBS supplemented with 10% FBS, and aliquot and store it at -80 °C.
- Determine the HBV DNA copy number present in the supernatant by HBV DNA qPCR (step 6).
5. Infection of 3D Cultures with HBV
- Perform infections in a class II cabinet within a containment level III lab.
- 3 days after seeding the monocultures or cocultures, thaw the required number of HBV-containing aliquots at room temperature and dilute the required virus dose in 1.8 mL of hepatocyte maintenance medium or coculture maintenance medium II per well, respectively.
- This 1.8-mL diluted virus is sufficient for the washing step (400 µL) and the replacement of medium in the well (1.4 mL). However, the required multiplicity of infection needs to be adjusted to account for the final culture volume of 1.6 mL.
- Transfer the plate to the washing dock and aspirate all medium from the wells. Add 400 µL of HBV-containing medium and initiate flow in the downward direction at 1 µL/s for 3.5 min. Aspirate all medium appearing on the reservoir side of the wells.
- Add 1.4 mL of HBV-containing maintenance medium/coculture maintenance medium II per well and transfer the plate into the docking station within a humidified incubator at 37 °C and with 5% CO2. Initiate flow in the downward direction at a flow rate of 1 µL/s for 8 h, followed by a reversal to the upward direction at a flow rate of 1 µL/s.
- 24 h following the addition of HBV, transfer the plate to the washing dock and aspirate all medium from the wells.
- Wash each well in the plate 3x with the corresponding medium, according to the type of culture as outlined in steps 2.12–2.14, to eliminate leftover virus from the well. In contrast to steps 2.12–2.14, add 1.6 mL of medium in every well to account for the extra volume to be sampled to exclude inoculum carryover (Figure 1g).
- Following these washing steps, collect 200 µL of medium from each well to confirm the complete removal of the HBV inoculum by quantification of extracellular HBV DNA.
- Transfer the plate to the docking station within a humidified incubator at 37 °C and with 5% CO2, and initiate flow in the upward direction at a flow rate of 1 µL/s for 48 h.
- 48 h later, collect the complete well volume for downstream analysis, followed by three washes with hepatocyte maintenance medium as outlined in steps 2.12–2.14. Replace the medium and wash each well 3x every 48 h until experimental termination.
6. Quantification of Extracellular HBV DNA
- Isolate total DNA from the culture supernatants according to the manufacturer's instructions with the addition of 1 µg of carrier RNA in a containment level III lab to ensure the virus inactivation in the samples prior to moving them to a different area.
- Prepare a master mix containing quantitative PCR master mix, 600 nM forward primer, 600 nM reverse primer, and 300 nM of probe.
- Add 7 µL of the master mix into each well of a 384-well plate.
- Add 5 µL of DNA samples in duplicate, a no-template control, and duplicates of serially diluted HBV genome-containing plasmid-based standard (e.g., pCMV-HBV) ranging from 109 copies per reaction to 102 copies per reaction to each well of the qPCR plate.
- Place an adhesive cover over the plate and ensure that each well is sealed correctly.
- Centrifuge the plate for 1 min at 300 x g.
- Start the qPCR run according to the manufacturer's instructions. The cycle conditions for real-time PCR are 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min.
- Quantify the number of HBV DNA copies within the unknown samples according to the standard curve.
7. Quantification of Intracellular HBV Pregenomic (pg)RNA
- Isolate total RNA from the scaffolds according to the manufacturer's instructions. In order to ensure complete cell lysis, vortex each scaffold 3x for 30 s followed by centrifugation at 300 x g for 1 min between each vortexing. Perform the cell lysis in the containment level III lab to ensure the virus inactivation in the samples prior to moving them to a different area.
- Transcribe cDNA from the isolated RNA according to the manufacturer's instructions.
- The cycle conditions for the retrotranscription are 25 °C for 10 min, 37 °C for 120 min, and 85 °C for 5 min.
- Keep the cDNA samples at 4 °C for short-term or at -20 °C for long-term storage.
- Prepare master mixes for pgRNA and RPS11 containing quantitative PCR master mix and forward and reverse primers for pgRNA and RPS11 (used as housekeeping gene) at a final concentration of 0.2 µM.
- Add 7.5 µL of the master mix and 2.5 µL of cDNA per well on a 384-well plate. Measure RPS11 and pgRNA of each sample in duplicate and include a no-template control well for both genes.
- Place an adhesive cover over the plate and ensure that each well is sealed completely.
- Centrifuge the plate for 1 min at 300 x g.
- Insert the plate into the qPCR cycler and start the qPCR run using the standard quantitative PCR protocol according to the manufacturer's instructions. The cycle conditions for real-time PCR are 50 °C for 2 min, 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min.
- Calculate the expression of pgRNA relative to RPS11.
8. Immunofluorescence Staining of Viral Antigen
- Remove the retaining ring from the well and remove the scaffold with forceps in a class II cabinet in the containment level III lab.
- Fix the cell-containing scaffolds with 4% paraformaldehyde in 1 mL of PBS for 30 min at room temperature in the containment level III lab. The following steps can be performed in a different area.
- Wash the scaffolds 3x with 1 mL of PBS.
- Permeabilize the cells using 0.1% Triton-X 100 in 1 mL of PBS for 1 h at room temperature.
- Wash the scaffolds 3x with 1 mL of PBS.
- Block non-specific binding by incubation of the scaffolds with 1% BSA in 1 mL of PBS for 16 h at 4 °C.
- Wash the scaffolds 3x with 1 mL of PBS.
- Perform primary antibody staining using rabbit anti-hepatitis B virus core antigen at a dilution of 1:200 in 1% BSA in 1 mL of PBS for 16 h at 4 °C.
- Wash the scaffolds 1x with 0.1% Tween in 1 mL of PBS (PBS-Tween) and 3x with 1 mL of PBS.
- Perform secondary antibody staining using goat anti-rabbit IgG (H+L) cross-adsorbed Alexa Fluor 594-conjugated secondary antibody at a dilution of 1:2,000 in 1% BSA in 1 mL of PBS for 16 h at 4 °C.
- Wash the scaffolds 1x with 1 mL of 0.1% PBS-Tween and 3x with 1 mL of PBS.
- Counterstain the scaffolds using DAPI in 1 mL of PBS at a concentration of 2 µg/mL for 10 min at room temperature.
- Wash the scaffolds 1x with 1 mL of 0.1% PBS-Tween and 3x with 1 mL of PBS.
- Transfer the scaffolds to a microscope slide and mount it for imaging.
- Image the scaffolds using a fluorescence microscope.
9. Human Albumin ELISA
- Perform this section of the protocol in a class II cabinet allocated in the containment level III lab if working with infectious material.
- To evaluate the viability and metabolic functionality of PHH, evaluate human albumin production by ELISA.
- Coat 96-well plates with 50 µL per well of goat anti-human antibody diluted 1:800 in coating buffer (100 mM bicarbonate/carbonate, pH 9.6). Cover the plates and incubate for 2 h at 37 °C or overnight at 4 °C.
- Aspirate the coating antibody from the plate and wash it 4x with 200 µL of 0.05% PBS-Tween.
- Add 200 µL of blocking buffer (1% BSA in PBS), cover the plates, and incubate them for 1 h at 37 °C or store them at 4 °C for 3 months. For long-term storage, add 0.05% sodium azide to the blocking buffer.
- Aspirate the blocking buffer and wash 1x with 200 µL of 0.05% PBS-Tween.
- Add 50 µL of previously diluted samples per well (1:100). The sample diluent contains 1% BSA in 0.05% PBS-Tween. Incubate for 1 h at 37 °C or overnight at 4 °C.
NOTE: Incubate the standards at the same time as the samples. A concentration range of 500–0.488 ng/mL (1:2 serial dilutions) is recommended. Perform all serial dilutions of human albumin in sample diluent. - Aspirate the samples from the plate and wash it 4x with 200 µL of 0.05% PBS-Tween.
- Add 50 µL of HRP-conjugated goat anti-human albumin antibody previously diluted 1:10,000 in sample diluent. Incubate for 2 h at 37 °C or overnight at 4 °C.
- Aspirate the antibody from the plate and wash it 6x with 200 µL of 0.05% PBS-Tween.
- Add 100 µL of TMB reagent and, as soon as the highest standards are fully developed, add 100 µL 1 M H2SO4 to stop the colorimetric reaction.
- Read the absorbance at 450 nm on a 96-well plate reader for analysis.
10. Interleukin (IL)6 and Tumor Necrosis Factors (TNF)α Production in 3D Co-cultures
- Perform this section of the protocol in a class II cabinet allocated in the containment level III lab if working with infectious material.
- Quantify the IL6 and TNFα production to evaluate the functionality and viability of the primary Kupffer cells. To induce the production of these cytokines by Kupffer cells, treat cocultures with 1 µg/mL lipopolysaccharide (LPS) 9 d post-seeding in the coculture maintenance medium II for 48 h.
- At day 11 post-seeding, harvest the medium from each well and store it at -80 °C.
- Measure the IL6 and TNFα concentration in the culture medium by an appropriate assay and according to the manufacturer's instructions.
Representative Results
We describe a simple and versatile platform for the long-term culture of primary human Kupffer cells and/or hepatocytes and their infection with HBV. Primary human cells are seeded on collagen-coated polystyrene scaffolds within a microfluidic plate assembly, which continuously perfuses the cells with growth medium (Figure 1a).
PHH, which usually are only stable for a limited amount of time in conventional culture systems, can be functionally maintained for extended periods of time. Human albumin, which is secreted by functional hepatocytes and is considered the best marker for the evaluation of hepatic metabolism, is stably and highly expressed by 3D cultures until day 40 post-seeding (Figure 2). For cocultures, Kupffer cell functionality and viability can be evaluated by the secretion of specific cytokines (e.g., IL6 and TNFα). To measure cytokine production, the use of capture-based detection means upon LPS-stimulation of cocultures is recommended (Figure 3).
Cells form hepatic microtissues, usually within 3 days of seeding of PHH, demonstrating functional bile canaliculi and complete cell polarization (Figure 2). In addition to retaining their physiological cellular metabolism, these cultures become exceptionally susceptible to HBV infection. HBV DNA and other viral markers, in contrast to other culture systems, become readily detectable from day 2 post-infection (Figure 4). In addition to secreted markers of viral infection, hepatocyte-containing scaffolds can be retrieved from the cultures and used for the immunofluorescence detection of viral antigens (e.g., HBsAg, HBcAg) (Figure 4). Where conventional hepatocyte cultures require inoculation with at least 500 HBV GE per cell and the addition of 2% DMSO and 4% PEG, as few as 0.05 HBV GE are able to initiate infection in 3-D cultures without the requirement of DMSO or PEG (Figure 4).
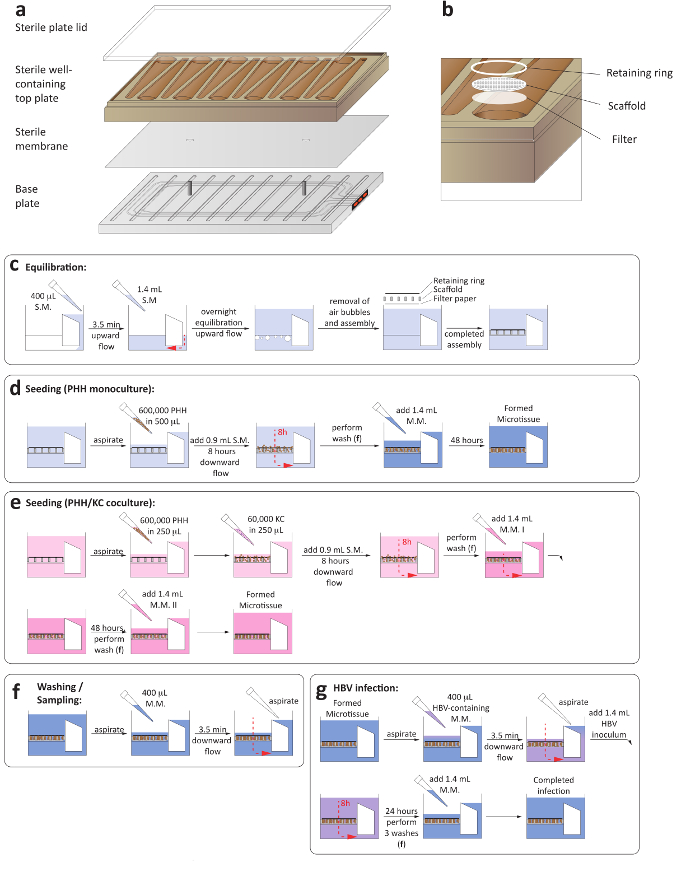
Figure 1: Set-up of 3-D liver-on-a-chip cultures. (a) This is a schematic layout for the assembly of the culture plate in order to ensure the establishment of microfluidic circulation. (b) This panel shows a close-up view of the culture wells, including the filter paper, scaffold, and retaining ring. (c) This panel shows the process of plate equilibration prior to seeding the cultures. The next two panels show the process of seeding for (d) hepatocyte monocultures and (e) hepatocyte/Kupffer cell cocultures. (f) This panel shows the washing steps involved in medium changes. (g) This panel shows the HBV infection set-up, including the removal of inoculum. S.M. = seeding medium, M.M. = maintenance medium. Please click here to view a larger version of this figure.
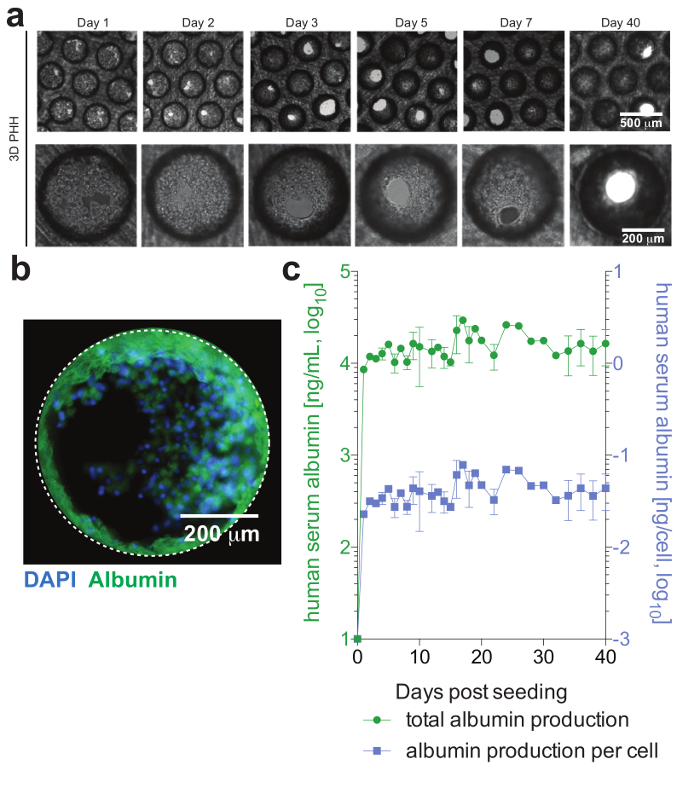
Figure 2: Hepatic microtissue formation and hepatocyte viability. (a) This panel shows longitudinal brightfield images of 3D hepatocyte monocultures demonstrating microtissue formation following seeding. (b) This panel shows immunofluorescence imaging of cultures for nuclei (blue) and human albumin (green). (c) This panel shows longitudinal total albumin, as well as per cell adjusted albumin production, during 40 days of hepatocyte monocultures, as determined by ELISA. The data shown are mean ± SD. This figure is adapted from Ortega-Prieto et al.4. Please click here to view a larger version of this figure.
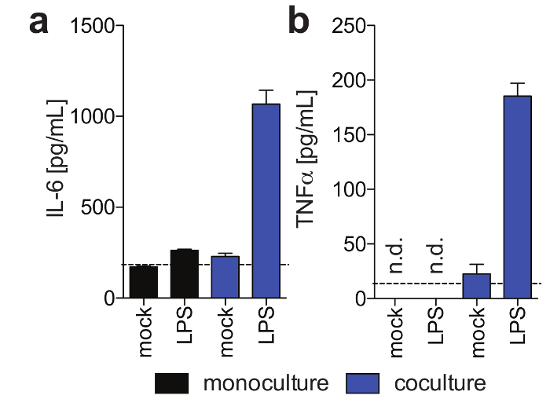
Figure 3: Kupffer cell functionality in 3-D cocultures. These panels show the secretion of (a) IL6 and (b) TNFα in hepatocyte monocultures and hepatocyte/Kupffer cell cocultures 11 days post-seeding in response to exogenously added LPS at day 9 post seeding, as determined using Human Magnetic Luminex assay. This figure is adapted from Ortega-Prieto et al.4. Please click here to view a larger version of this figure.
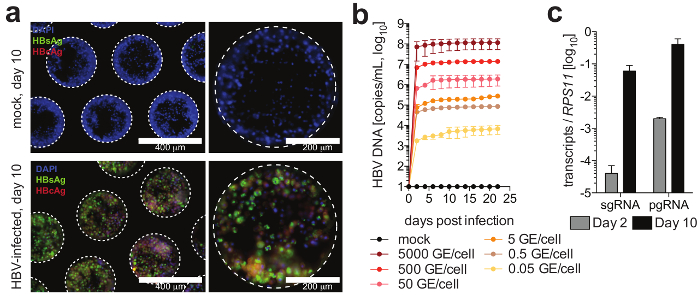
Figure 4: HBV infection in liver-on-a-chip cultures. (a) This panel shows the immunofluorescence microscopy detection of HBcAg (red), HBsAg (green), and nuclei (blue) 10 days following the infection of the cultures with HBV. (b) This panel shows the susceptibility of the cultures to HBV infection using different multiplicities of infection, as determined by a quantification of HBV DNA in the culture supernatants. (c) This panel shows the quantification of the longitudinal accumulation of HBV pgRNA relative to the housekeeping gene RPS11. The data shown are mean ± SD. This figure is adapted from Ortega-Prieto et al.4. Please click here to view a larger version of this figure.
Discussion
The challenges in maintaining long-term cultures of PHH have driven the development of several culture models with increased functionality and longevity, each exhibiting differential advantages and disadvantages. It is now widely acknowledged that static 2D cultures of PHH are mimicking certain aspects of hepatocyte biology for very limited amounts of time. Thus, micropatterned cocultures12,13, spheroid cultures14,15, and 3D liver-on-a-chip cultures16,17 are rapidly replacing these more basic systems. Especially when studying infectious diseases, which have coevolved with their host to utilize specific microenvironments, the requirement for providing physiological environments is underpinned by the often challenging nature of culturing human-tropic infectious diseases, including hepatitis C virus, HBV, and malaria.
The most critical step in performing 3D liver-on-a-chip cultures is the quality of the initially sourced primary cell types. These cells should be tested for their adherence capacity and only plateable PHH lots should be used in order to ensure successful tissue formation and culture generation. Even though freshly isolated PHH can be used, their cryopreservation is usually complicated and requires special rate-controlled freezers.
In contrast to conventional static 2D cultures, the host genetic background is negligible in regard to susceptibility to HBV infection, and all thus-far tested hepatocyte donors are able to establish HBV infection4.
Even though patient-derived HBV establishes infections of 3D cultures, it is imperative to utilize PEG-precipitated and sucrose cushion-purified HBV whenever using inducible HBV producer cell lines for the generation of viral inocula. Cell culture supernatants directly applied to 3D liver-on-a-chip cultures, either through the presence of inhibitory factors or due to an incompatibility of present growth factors with hepatocytes, do not readily result in infection. Additionally, when selecting patient-derived viral inocula, only serum should be used, since plasma inevitably coagulates and clogs the microfluidic circulation of the culture platform.
Irrespective of the viral inoculum used, assuring cellular viability and differentiation, as well as ensuring complete removal of the initial HBV inoculum, is key to successful long-term infection studies. The most convenient way to do this is sampling cultures following the removal of the viral inoculum, as well as measuring human serum albumin levels throughout the culture period. Of note, similarly to all other described platforms, HBV infection, once established, does not readily spread to uninfected cells. The underlying mechanism for this remains elusive since HBV infection in vivo readily infects the majority of the hepatocytes within the liver.
In regard to cocultures of PHH and Kupffer cells, it is advisable to perform lot tests of Kupffer cells to evaluate IL6 and TNFα secretion in response to LPS stimulation, since not all commercially available Kupffer cell donors have an equal responsiveness.
Importantly, for all drug treatments or initial infection of cultures with HBV, the total volume of the well (1.4 mL), as well as of the microfluidic channel (0.2 mL), must be taken into account for the calculation of drug or inoculum concentrations. In order to assure accurate dosing, one washing step with medium containing HBV or drugs is performed in order to prime the microfluidic channel.
The platform used utilizes 600,000 PHH per well, which ensures multilayered hepatocytes within the scaffolds. Even though the cell number can be varied, the chosen cell concentration ensures optimal results. The plate format holds a total of 12 scaffolds, which can be upgraded to 36 scaffolds. However, due to microfluidic requirements, scaling up to higher well numbers is not possible to date.
Using these approaches, cultures can be maintained with optimal cell performance for at least 40 days, which, thus far, offers unprecedented opportunities to evaluate novel drug candidates, as well as study the complex interplay between different hepatic cell populations during HBV infection.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
This work was funded by a Starter grant from the European Research Council (637304), a Wellcome Trust Investigator Award (104771/Z/14/Z), and by CN Bio Innovations.
Materials
| Reagents | |||
| William's E Medium, no phenol red | GIBCO | A12176-01 | |
| Hepatocyte Thaw Medium | GIBCO | CM7500 | |
| Primary Hepatocyte Thawing and Plating Supplements | GIBCO | CM3000 | |
| Primary Hepatocyte Maintenance Supplements | GIBCO | CM4000 | |
| DMEM/F-12 | GIBCO | 11320-033 | |
| Advanced DMEM | GIBCO | 12491023 | |
| DPBS, no calcium, no magnesium | GIBCO | 14190-144 | |
| MEM Non-Essential Amino Acids (NEAA) 100X | GIBCO | 11140050 | |
| Penicillin-Streptomycin (10,000 U/mL) | GIBCO | 15140-122 | |
| Fetal Bovine Serum, USA origin, Heat Inactivated, sterile-filtered, suitable for cell culture | SIGMA | 12106C | |
| Hydrocortisone | SIGMA | H0888 | |
| Trypan blue | Merck | T8154 | |
| Collagen from calf skin | Merck | C9791 | |
| G418 | SIGMA | G418-RO | |
| Tetracycline | SIGMA | T3258 | |
| Polyethylene glycol 8000 | SIGMA | P2139 | |
| Sucrose | SIGMA | SO389 | |
| Sodium carbonate anhydrous | SIGMA | 451614-25G | |
| Sodium bicarbonate | SIGMA | S5761 | |
| Sodium azide | SIGMA | S2002-5G | |
| Sulfuric acid, 99.999% | SIGMA | 339741 | |
| 4% Paraformaldehyde | SIGMA | 252549 | |
| Triton-X 100 | SIGMA | X100 | |
| Tween 20 | SIGMA | P1379 | |
| DAPI | SIGMA | D9564 | |
| Albumin (human) | SIGMA | A9731 | |
| Fisher BioReagent Bovine Serum Albumin, Fraction V, Heat Shock Treated | Fisher Scientific | BP9701-100 | |
| ProLong Gold Antifade Mountant | Invitrogen | P36930 | |
| TaqMan Universal Master Mix II, no UNG | Applied Biosystems | 4440040 | |
| SYBR Select Master Mix | Applied Biosystems | 4472903 | |
| Lipopolysaccharide from Escherichia coli K12 | InvivoGen | tlrl-eklps | |
| Name | Company | Catalog Number | Yorumlar |
| Kits/Consumables | |||
| Sterile membrane | CN Bio innovations | LC-SC | |
| LiverChip Perfusion cell culture plate | CN Bio innovations | LC12 | |
| LiverChip culture plate lid | CN Bio innovations | LC-SC | |
| Sterile round filter paper | CN Bio innovations | LC-SC | |
| Cell attachment scaffold | CN Bio innovations | LC-SC | |
| Retaining ring | CN Bio innovations | LC-SC | |
| Sterile plunger | CN Bio innovations | LC-ST | |
| Dneasy blood & tissue kit | Qiagen | 69506 | |
| Rneasy mini kit | Qiagen | 74106 | |
| Human Magnetic Luminex assay | R&D Systems | ||
| 1-Step Ultra TMB-ELISA Substrate Solution | ThermoFisher Scientific | 34028 | |
| High Capacity cDNA Reverse Transcription Kit | ThermoFisher Scientific | 4368814 | |
| QIAamp Viral RNA Mini Accessory Set | Qiagen | 1048147 | Containing RNA carrier |
| Millicell HY 5-layer cell culture flask, T-1000, sterile | Millipore (Merck) | PFHYS1008 | |
| MicroAmp Optical 384-Well Reaction Plate with Barcode | Life technologies | 4309849 | |
| MicroAmp Optical Adhesive Film | Life technologies | 4311971 | |
| Clear Flat-Bottom Immuno Nonsterile 96-Well Plates | ThermoFisher Scientific | 442404 | |
| Sealing Tape for 96-Well Plates | ThermoFisher Scientific | 15036 | |
| Nalgene Rapid-Flow Sterile Disposable Bottle Top Filters with PES Membrane | ThermoFisher Scientific | 295-3345 | |
| Fisherbrand Microscopic Slides with Ground Edges, Twin Frost | Fisher Scientific | FB58628 | |
| Tube, Thinwall, Ultra-Clear, 38.5 mL, 25 x 89 mm | Beckman Coulter | 344058 | |
| Name | Company | Catalog Number | Yorumlar |
| Primary cells / Cell lines | |||
| Human Plateable Hepatocytes, Transporter Qualified | Thermo Fisher Scientific | HMCPTS | |
| Cryopreserved Human Kupffer Cells | Thermo Fisher Scientific | HUKCCS | |
| HepDE19 cell line | Haitao Guo (Indiana University, IN, USA) | ||
| Name | Company | Catalog Number | Yorumlar |
| Primers/Probes/Standards | |||
| HBV DNA forward primer | Invitrogen | 5'-GTGTCTGCGGCGTTTTATCA-3' | |
| HBV DNA reverse primer | Invitrogen | 5'-GACAAACGGGCAACATACCTT-3' | |
| HBV DNA probe | Invitrogen | 5'FAM-CCTCTKCATCCTGCTGCTATGCCTCATC-3'TAMRA | |
| pgRNA forward primer | Invitrogen | 5'-GAGTGTGGATTCGCACTCC-3' | |
| pgRNA reverse primer | Invitrogen | 5'-GAGGCGAGGGAGTTCTTCT-3' | |
| RPS11 forward primer | Invitrogen | 5'-GCCGAGACTATCTGCACTAC-3' | |
| RPS11 reverse primer | Invitrogen | 5'-ATGTCCAGCCTCAGAACTTC-3' | |
| pCMV-HBV | Professor Christoph Seeger (Fox Chase Cancer Centre, PA, USA) | ||
| Name | Company | Catalog Number | Yorumlar |
| Antibodies | |||
| Anti-Hepatitis B virus core antigen IgG fraction (polyclonal) | DAKO | discontinued | Lot 10102505 |
| Human Albumin Antibody, A80-129A | Bethyl Laboratories. inc | A80-129A | |
| Human Albumin cross-adsorbed Antibody, A80-229P | Bethyl Laboratories. inc | A80-229P | |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher Scientific | # A-11072 | Lot 1431810 |
| Name | Company | Catalog Number | Yorumlar |
| Equipment | |||
| LiverChip Vacuum pump | CN Bio innovations | LC-PN | |
| LiverChip Pneumatic Hookup | CN Bio innovations | LC-PN | |
| LiverChip platform | CN Bio innovations | LC-PN | |
| LiverChip plate washing dock | CN Bio innovations | ||
| Autoclavable metal forceps | VWR | 232-0106 | |
| Vortex Genie 2 | Scientific industries | SKU: SI-0236 | |
| Optima XPN-80 Ultracentrifuge | Beckman Coulter | A95765 | |
| Heraeus Multifuge X3R Centrifuge | Thermo Scientific | 75004500 | |
| SAM-12 Medical Suction High Vacuum High Flow | MGE worldwide | SAM12/01010101 | |
| NUAIRE 5800 SERIES incubator | NUAIRE | NU-5841 | |
| Automated precision torgue | CN Bio innovations | ||
| Manual torque | CN Bio innovations | ||
| LiverChip compressor | CN Bio innovations | ||
| Luminex LX-200 Instrument with xPONENT 3.1 | Luminex | ||
| Millipore Hand-Held Magnetic Separator Block | ThermoFisher Scientific | Millipore™ 40-285 | |
| FluoStar Optima Plate Reader | BMG Labtech | ||
| KOLVER Precision electric screwdrivers | VTECH ltd | FAB10RE/FR | |
| KOLVER Power supply | VTECH ltd | EDU1FR | |
| BAMBI VTS75D | Air Equipment | Discontinued | |
| Integra Vacuboy | INTEGRA | ||
| ViiA 7 Real-Time PCR System with 384-Well Block | ThermoFisher Scientific | 4453536 | |
Referanslar
- Verrier, E. R., Colpitts, C. C., Schuster, C., Zeisel, M. B., Baumert, T. F. Cell Culture Models for the Investigation of Hepatitis B and D Virus Infection. Viruses. 8 (9), (2016).
- Elaut, G., et al. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Current Drug Metabolism. 7 (6), 629-660 (2006).
- Konig, A., et al. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. Journal of Hepatology. 61 (4), 867-875 (2014).
- Ortega-Prieto, A. M., et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nature Communications. 9 (1), 682 (2018).
- Allweiss, L., Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses. 9 (6), (2017).
- Guo, J. T., Guo, H. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Research. 122, 91-100 (2015).
- Lucifora, J., et al. Direct antiviral properties of TLR ligands against HBV replication in immune-competent hepatocytes. Scientific Reports. 8 (1), 5390 (2018).
- Hosel, M., et al. Hepatitis B Virus Activates Signal Transducer and Activator of Transcription 3 Supporting Hepatocyte Survival and Virus Replication. Cellular and Molecular Gastroenterology and Hepatology. 4 (3), 339-363 (2017).
- Mazza, G., et al. Rapid production of human liver scaffolds for functional tissue engineering by high shear stress oscillation-decellularization. Scientific Reports. 7 (1), 5534 (2017).
- Hang, T. C., Lauffenburger, D. A., Griffith, L. G., Stolz, D. B. Lipids promote survival, proliferation, and maintenance of differentiation of rat liver sinusoidal endothelial cells in vitro. American Journal of Physiology-Gastrointestinal and Liver Physiology. 302 (3), G375-G388 (2012).
- Hwa, A. J., et al. Rat liver sinusoidal endothelial cells survive without exogenous VEGF in 3D perfused co-cultures with hepatocytes. The FASEB Journal. 21 (10), 2564-2579 (2007).
- Khetani, S. R., Bhatia, S. N. Microscale culture of human liver cells for drug development. Nature Biotechnology. 26 (1), 120-126 (2008).
- March, S., et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nature Protocols. 10 (12), 2027-2053 (2015).
- Bell, C. C., et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Scientific Reports. 6, 25187 (2016).
- Tong, W. H., et al. Constrained spheroids for prolonged hepatocyte culture. Biomaterials. 80, 106-120 (2016).
- Griffith, L. G., Wells, A., Stolz, D. B. Engineering liver. Hepatology. 60 (4), 1426-1434 (2014).
- Domansky, K., et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 10 (1), 51-58 (2010).
