When performing metabolic labeling of newly transcribed RNA, several aspects need to be controlled: the time and efficiency of the labeling, the spike-in proportion, the extraction protocol, and the biotinylation efficacy (including signal-to-noise ratio), among others. These conditions have been extensively and methodically shown by others7,10,11. Here we mainly focus on the interpretation and immediate analyses that can be performed once the samples have been processed, either by RT-qPCR, microarray, or sequencing. The analyses of different mutant strains demonstrate the power of the method to detect not only a dramatic global decrease in mRNA synthesis, as in the case of SAGA mutant S. cerevisiae strains, but also a very mild reduction of RNA polymerase II activity upon suppression of histone H2B monoubiquitination.
Our analyses of SAGA enzymatic activities suggested a broad recruitment to chromatin15, which was not revealed by the analysis of steady-state mRNA levels in SAGA mutant strains. As RNA polymerase II recruitment was impaired upon SAGA inactivation, we decided to analyze whether mRNA synthesis rates would be globally affected. Hence, wild-type or mutant S. cerevisiae strains were exposed to 4tU for a 6 min period, to label newly transcribed RNAs. After mixing with the spike-in labeled cells (S. pombe) in a proportion of 3:1, total RNA was extracted, and newly synthesized RNA was biotinylated and purified according to the protocol presented here, as in the chronogram shown in Figure 1. Labeled RNAs were purified from a total of 200 µg of RNA, ensuring that the amount of purified product would be sufficient for any downstream application. As an initial and systematic step before any genome-wide analysis, newly synthesized RNA purification was validated by RT-qPCR. Genes were selected according to different parameters, including a level of expression, regulatory pathways, and a dependence on different RNA polymerases.
To confirm that this protocol specifically purifies labeled RNA, we quantified the levels of transcripts in fractions purified from wild-type cells that were cultured with or without 4tU. Negligible background levels of the analyzed RNAs were detected from cells that were not exposed to 4tU (Figure 2A). Newly transcribed RNA purification was further validated by the observed enrichment of the intron-containing ACT1 pre-mRNA (data not shown). After validation of the quality of the samples, we tested whether mRNA synthesis would be affected upon deletion of SPT20, which is known to disrupt the SAGA complex assembly. As reported by others, mRNA quantification performed on total (steady-state) RNA from the spt20Δ strain revealed mostly unchanged or mildly reduced levels for the tested genes (Figure 2B). Similar results were obtained for strains deleted for BRE1 (Figure 2B). In contrast, the analysis of newly transcribed RNA from the spt20Δ strain revealed a dramatic decrease in mRNA synthesis by three- to fivefold for all tested genes (Figure 2C). The loss of Bre1 led to a more discreet but still visible decrease in newly synthesized mRNA levels for the studied genes (Figure 2C). In good agreement with a role of SAGA and Bre1 in RNA polymerase II transcription, the loss of either Spt20 or Bre1 did not affect the expression of RDN25 or snR6 genes, transcribed by RNA polymerase I and RNA polymerase III, respectively (Figure 2C).
However, strains deleted for structural subunits of the SAGA complex, like SPT7 or SPT20, display severe slow-growth phenotypes which may account for the observed transcriptional alterations. To rule out undesired secondary effects, we conditionally depleted Spt7 from the nucleus, using anchor-away S. cerevisiae strains14,24. Upon 60 min of rapamycin treatment, prior to the pulse-labeling with 4tU (see Figure 1 for a schematic representation), newly transcribed mRNA levels were reduced to a similar extent as that observed in the deletion strain (Figure 2D and 2E). This analysis, thus, confirmed our former results and validated the protocol for this inducible depletion system. In a time-course analysis, where cells were exposed to rapamycin for a time spanning from 0 to 240 min, reduced expression was evidenced immediately after 15 min of exposure to the drug. More interestingly, steady-state mRNA levels tended to initially diminish but returned to normal levels after 60 min, an indication that a compensatory mechanism takes place in the meantime (Figure 2E).
Altogether, the labeling and quantification of newly synthesized RNA allowed revealing new regulatory roles for the SAGA complex. The described protocol could also reveal moderate effects on RNA polymerase II activity and was successfully applied to conditional depletion yeast strains.
One of the downstream applications for the purified newly synthesized RNA is a genome-wide quantification of transcripts using microarray hybridization or sequencing (4tU-seq). While high-throughput sequencing is more quantitative, sensitive and informative, microarray hybridization can be very helpful to determine whether the global mRNA levels are altered. In this context where normalization is pivotal, we added a spike-in organism to the sample that we aimed to analyze. Specifically, we mixed S. cerevisiae cells to S. pombe cells in a ratio of 3:1, both being previously exposed to 4tU. When the purified, labeled RNAs are subjected to high-throughput sequencing, any standard library preparation protocol can be used, most often following ribosomal RNA depletion. Normalization between 4tU-seq data from different samples has been performed by adding either labeled RNA from a different species (i.e., mixing S. cerevisiae and S. pombe cells as above)22 or in vitro-transcribed, thiolated spike-in RNA12,25,26,27. Commercially available microarray chips contain probes for the whole transcriptome of both budding and fission yeast, allowing the quantification of mRNA from both organisms in one single experiment. Microarray hybridizations were performed with total and newly synthesized RNA validated by RT-qPCR as indicated above (wild-type, spt20Δ, and bre1Δ). In addition, we included a strain that does not support monoubiquitination of histone H2B, through point mutation of the ubiquitinable residue (K123R). Because the exact same proportion of S. pombe cells were used in the different samples and replicates, it is possible to linearly rescale the arrays' intensities so that the total and labeled S. pombe probes have the same median intensity. This normalization, or rescaling, can be performed using a Bioconductor package developed by the laboratory of Patrick Cramer and is used in parallel in all analyzed samples, total and labeled fractions and wild-type and mutants strains8. Briefly, the input of this pipeline is an Excel file containing the probes and their intensity (MAS5 or RMA) for all the samples and fractions. After excluding probes that are outside the range of detection, the signal intensity is rescaled, taking into account the intensity values of the S. pombe probes. Finally, you can map the probes to the budding and fission yeast genomes, ending with a matrix containing the expression values normalized for the spike-in. These values can be further processed (see next section) or used as is. In the following example, we determined the fold change of every transcript between the mutant and the wild-type strain.
The analyses were performed for both steady-state or newly synthesized levels of RNA and plotted against their statistical significance (p-value) (Figure 3). In agreement with other studies, when total RNA levels were analyzed, only a few genes had their expression altered, either up- or downregulated (Figure 3A-3C). However, the analysis of newly transcribed RNA led to strikingly different conclusions. The newly transcribed mRNA levels of more than 4,000 genes were significantly reduced by at least twofold upon deletion of SPT20, suggesting a global positive effect of SAGA on RNA polymerase II transcription in budding yeasts (Figure 3D). Additionally, in the bre1Δ and K123R mutants, the results were more discreet: most genes appeared to have their expression reduced, but the extent of downregulation and the number of significantly affected genes (≈ 300 – 500) was indeed more limited (Figure 3E and 3F).
As previously mentioned, steady-state or total levels of mRNA are dictated by the tight equilibrium between synthesis and decay (Figure 4A). When RNA polymerase II transcription is globally impaired, two scenarios can be pictured: (i) either the total mRNA levels decrease globally, as a response to reduced synthesis but constant decay, or (ii) mRNA degradation is decreased to the same extent, resulting in mostly unchanged steady-state mRNA levels. The second scenario has been reported for several conditions, including in the context of SAGA or TFIID disruption9,10,14,16,22. A procedure called comparative dynamic transcriptome analysis (cDTA) based on 4tU labeling and dynamic kinetic modeling allows us to determine mRNA synthesis and infer the decay rates for every transcript8,10. Once again, we took advantage of the data collected for the previously mentioned strains (wild-type, spt20Δ, bre1Δ, and K123R). As expected, upon deletion of SPT20, we observed a simultaneous decrease in mRNA synthesis and degradation rates when compared to the wild-type strain. In this mutant strain, the compensation was almost optimal, with an average decrease in synthesis of 3.8-fold and an average decrease in decay of 4.1-fold (Figure 4B), corroborating why only limited transcriptional changes could be detected on total mRNA levels (Figure 3A). In the two other mutant strains (bre1Δ and K123R), concomitant changes in mRNA synthesis and decay were also observed, but the changes were on a much smaller scale and more dispersed (Figure 4C and 4D).

Figure 1: Schematic representation of the metabolic labeling of RNA using 4tU. Freshly prepared 4tU is added to the culture medium and cells are labeled for 6 min. Labeled S. cerevisiae and S. pombe cells are mixed in a ratio of 3:1 and total RNA is extracted. Afterward, newly synthesized RNA is biotinylated and can be purified using streptavidin-coated magnetic beads. Finally, total (steady-state) RNA and labeled newly synthesized RNA can be used in a variety of downstream applications, including RT-qPCR and microarray hybridization or sequencing. Please click here to view a larger version of this figure.
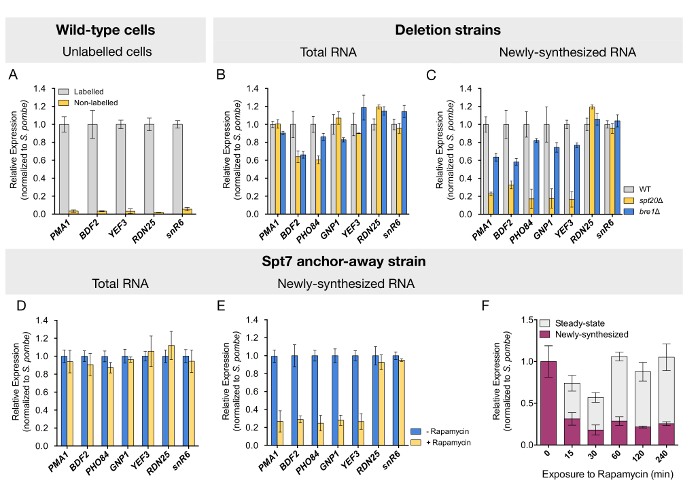
Figure 2: Analysis depicting transcriptional changes in both steady-state and newly transcribed RNA determined by RT-qPCR. (A) RNA levels of five different genes were quantified from the labeled RNA fraction purified from wild-type cells that were either exposed or not to 4tU. The next two panels show (B) total and (C) newly synthesized RNA quantification by RT-qPCR for wild-type (WT), spt20Δ, and bre1Δ yeast cells. Spt7 anchor-away strains, untreated or treated with rapamycin for 60 min, were labeled with 4tU, and (D) total or (E) newly transcribed RNA was quantified by RT-qPCR. (F) This panel shows the time-course analysis of changes in steady-state and newly synthesized mRNA upon Spt7 nuclear depletion. For all samples, mRNA levels for five RNA polymerase II genes were quantified by RT-qPCR. RNA polymerase I and RNA polymerase III genes (RDN25 and snR6, respectively) were used as control. Expression values (mean ± SD of three independent experiments) were normalized to the spiked-in S. pombe signal and set to 1 in the control sample. Panels D-F have been modified from Baptista et al.14. Please click here to view a larger version of this figure.
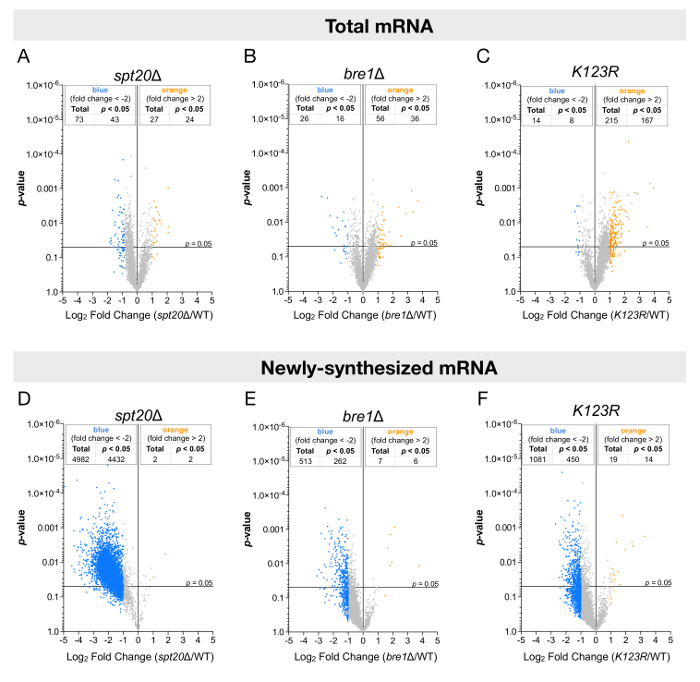
Figure 3: Genome-wide analyses of mRNA levels using total or labeled RNA fractions. These panels show volcano plots showing fold changes in (A-C) steady-state mRNA levels or (D-F) newly synthesized mRNA levels relative to their significance (p-value). The fold changes (FC) were calculated as the log2 of the ratio of the expression value of each gene after normalization to the S. pombe signal in the (A and D) spt20Δ, (B and E) bre1Δ, or (C and F) K123R strain versus the expression value of the same gene in wild-type S. cerevisiae. A total of 5,385 genes were analyzed, and thresholds of twofold change (blue dot: more than a twofold decrease; yellow dots: more than a twofold increase) and 0.05 p-values were considered. Panels A and D have been modified from Baptista et al.14. Please click here to view a larger version of this figure.
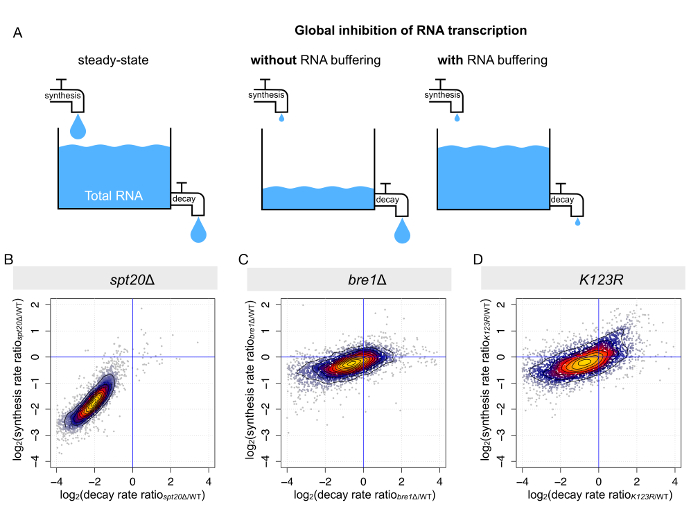
Figure 4: Parallel changes in mRNA synthesis and mRNA decay results in mRNA buffering. (A) This panel shows a schematic representation of the outcome of RNA synthesis perturbation on steady-state RNA levels. (B-D) These panels show the calculation of mRNA synthesis and the decay rates from the analyses of total and newly synthesized RNA. The synthesis and decay rates were determined for each S. cerevisiae transcript in (B) spt20Δ, (C) bre1Δ, and (D) K123R. Changes (calculated as the log2 of the ratio between mutant and wild-type) in synthesis rates were plotted against changes in decay rates. Please click here to view a larger version of this figure.
