Figure 1 shows a schematic of the workflow described in the protocol. To determine the contributions of ER-bound enhancers near the estrogen-regulated gene MMP17, which has 3 binding sites nearby as defined by ChIP-seq (Figure 2A), guide RNAs were designed for each region. To design guide RNAs, a 600 – 900 bp window of sequence surrounding each ER binding site of interest was selected and put into a guide RNA design program. Resulting guide RNA sequences with 0-2 predicted off target sites were aligned to the human genome using BLAT. Four non-overlapping guide RNAs that spanned the region defined by ChIP-seq and DNaseI hypersensitivity were chosen for targeting (Figure 2B). Additional sequence (Table 1) was added to each end to facilitate downstream cloning and the resulting 59 nucleotide fragments were ordered. Upon arrival, guide RNAs were diluted and pooled by site, and a short PCR was performed to add homology regions prior to Gibson assembly. Figure 2C shows the expected guide RNA product after a short PCR using the "U6_internal" primers (Table 1), which will add 20 basepairs of sequence to each end of the 59 basepair guide RNA fragment, resulting in a ~100 basepair sequence. Following Gibson assembly, these guide RNA pools were transformed into bacteria and plasmid minipreps were prepared the following day. Figure 2D shows results from an enhancer dissection experiment, where multiple enhancers nearby MMP17 are targeted alone and in combination using Enhancer-i. Sites targeted by Enhancer-i are indicated with a black hexagon. Guide RNA plasmids targeting the indicated sites were transfected into an estrogen-deprived Ishikawa cell line stably expressing SID4X-dCas9-KRAB. Two days later, the media was changed and puromycin was added to enrich for transfected cells. The following day, the cells were harvested following an 8 h 10 nM estradiol treatment. RNA was isolated, and a one-step qPCR was performed. In this example, sites 1 and 2 are necessary for a complete estrogenic response of MMP17, while site 3 does not contribute under these conditions (Figure 2D, lanes ii-iv). When only sites 2 or 3 are active (vi and vii), the estrogen response is similar to when no sites are active (viii), suggesting that these sites cannot contribute independently. Site 1 can contribute some expression by itself (v), but the greatest activity is seen when sites 1 and 2 are active (iv).
To manipulate 10 enhancers near 4 different genes simultaneously (Figure 3A), complex pools of guide RNAs were generated containing 42 enhancer guides and 16 promoter guides. Guide RNA oligos were pooled before the initial guide RNA extension PCR (Step 3.3), and resulting PCR products were purified and combined with the empty puromycin U6 cloning vector using Gibson assembly. Following the Gibson assembly, multiple independent transformations were performed and plated. The plates were scraped into LB and allowed to grow out for 2 – 4 h prior to maxiprep. Figure 3B shows representative reductions in gene expression by qPCR when these guide RNA pools were transfected into an estrogen-deprived Ishikawa cell line stably expressing SID4X-dCas9-KRAB and treated as described above (Figure 2D). Reductions from Enhancer-i are similar to those obtained by targeting the promoter of the putative target gene. Figure 3C shows the effects of dilution of guide RNAs on reduction of the estrogen response using Enhancer-i. A 1:50 dilution of a guide RNA pool targeting the enhancer near G0S2 still yields significant reduction in gene expression, suggesting that Enhancer-i can be used to target up to 50 sites at once. However, the deactivation can be diluted out, indicating that hundreds of sites cannot be targeted simultaneously unless more sensitive detection methods are employed.

Figure 1. Protocol schematic for multiplex enhancer dissection using Enhancer-i. Guide RNAs (red and blue) are designed using e-crisp and selected using the UCSC genome browser. Four guide RNAs are chosen that span the regions of interest (transcription factor binding sites as defined by ChIP-seq). Guide RNA oligonucleotides that have been pooled by the region of interest (red and blue) undergo a PCR to add homology regions (orange) prior to the Gibson assembly and transformation. Resulting plasmid pools are transfected via lipofection into cell lines stably expressing SID4X-dCas9-KRAB or into wild-type cells in conjunction with SID4X-dCas9-KRAB plasmid. Guide RNA plasmid pools can be transfected individually to target one site at a time, or in combination to target multiple sites simultaneously. Transfected cells are treated with antibiotics to enrich for cells containing guide RNAs. At ~72 h post transfection, the cells are harvested. Nucleic acids can be extracted for qPCR, RNA-seq, or ChIP-seq. Please click here to view a larger version of this figure.
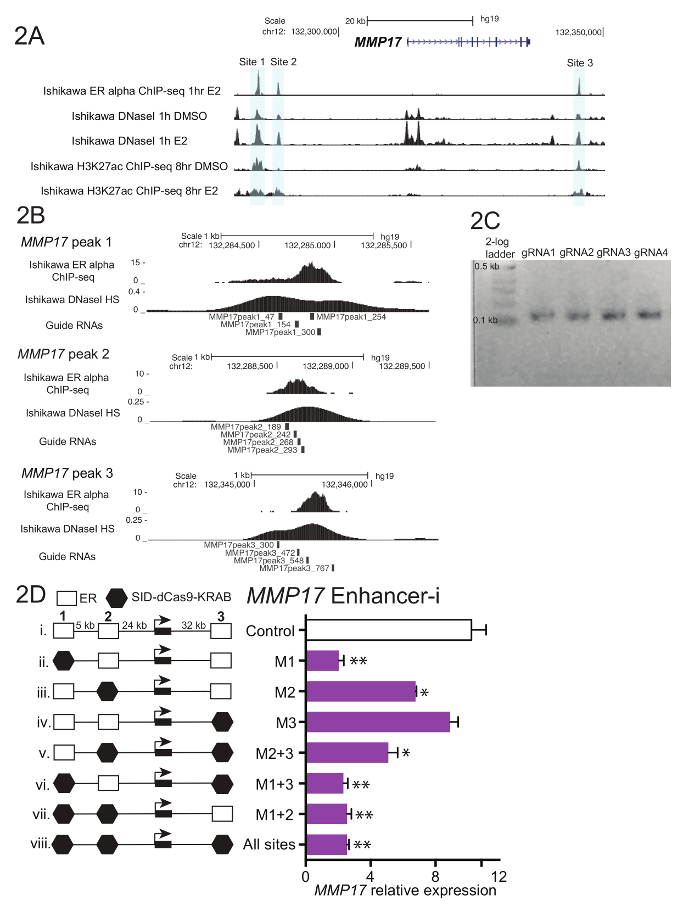
Figure 2. Guide RNA design and enhancer dissection for MMP17. (A) Genome browser screenshot of the ER alpha-bound enhancers (gray) to be targeted near MMP17. This figure has been modified from Carleton, et al.18. (B) Guide RNA designs for the 3 binding sites18. The binding site for ER as defined by ChIP-seq is the target, and the 4 guide RNAs tile across this region. The DNaseI sensitivity signal, which spans the binding site, can also be used to define target sequence for guide RNA design. Both ChIP-seq and DNaseI HS data were obtained from Ishikawa cells treated with 10 nM estradiol for 1 h. (C) Representative guide RNA sequences that are ready for Gibson assembly, having undergone a short PCR to add homology regions. (D) Relative expression of MMP17 measured via qPCR following targeting of specific regions with Enhancer-i and an 8-h10 nM estradiol treatment. Expression is relative to CTCF and expression level of MMP17 in cells not treated with estradiol. Control guide RNAs target the promoter of IL1RN. All error bars represent SEM, double asterisks indicate p <0.01 and single asterisks indicate p <0.05 in a paired t-test. This figure has been modified from Carleton, et al.18. Please click here to view a larger version of this figure.
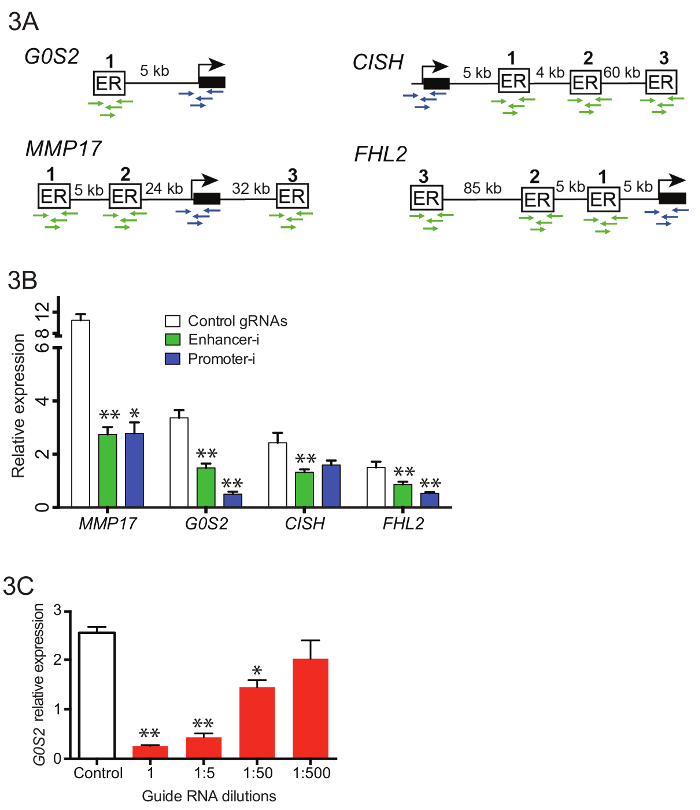
Figure 3. Targeting multiple enhancers near different genes simultaneously with pooled Enhancer-i. (A) Schematic of the binding sites and promoters to be targeted in pooled Enhancer-i. (B) The effects on expression as measured by qPCR after E2 treatment on Ishikawa cells transfected with Enhancer-i plasmid pool (green), Promoter-i plasmid pool (blue) or control gRNAs (white)18. A significant reduction at all genes is observed with Enhancer-i. This figure was modified from Carleton, et al.18. (C) The effects on G0S2 expression levels after E2 treatment on Ishikawa cells transfected with different amounts of guide RNAs targeting G0S2. A significant reduction can be seen even with small amounts of guide RNA (1:50 dilution), suggesting that up to 50 sites may targeted simultaneously. All error bars represent SEM, double asterisks indicate p <0.01 and single asterisks indicate p <0.05 in a paired t-test. Please click here to view a larger version of this figure.
| Name | Sequence |
| U6_internal_F | TTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCG |
| U6_internal_R | GACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC |
| U6_PCR_F | CCAATTCAGTCGACTGGATCCGGTA |
| U6_PCR_R | AAAAAAAGCACCGACTCGGTGCCA |
| gRNA_qPCR_F | GCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCG |
| gRNA_qPCR_R | AAAAAGCACCGACTCGGTGCC |
| dCas9_qPCR_F | GTGACCGAGGGAATGAGAAA |
| dCas9_qPCR_R | AGCTGCTTCACGGTCACTTT |
| pAC95_PCR_F | AGAAGAGAAAGGTGGAGGCC |
| pAC95_PCR_R | CGTCACCGCATGTTAGAAGG |
| SID4X_PCR_F | CAATAGAAACTGGGCTTGTCG |
| SID4X_PCR_R | TCGTGCTTCTTATCCTCTTCC |
Table 1. Primers used for guide RNA extension and sequencing, qPCR, and detection of the fusion protein.
