Using the protocols described here for the utilization of enzyme-catalyzed activation (i.e., P450, peroxidase, reductase) to investigate potency of chemicals (carcinogens/drugs) to be metabolized to intermediates resulting in their covalent binding to DNA (generation of DNA adducts), we were able to resolve (i) a novel mechanism of the pharmacological action of the anticancer agent ellipticine (for a review see,29,30,31), (ii) the etiology of two nephropathies associated with upper urothelial tract cancer caused by plant alkaloid aristolochic acid (Aristolochic acid nephropathy and Balkan endemic nephropathy) (for a review see,32,33,34,35,36,37,38,39), and (iii) the genotoxic mechanisms of carcinogenicity of several carcinogens such as an air pollutant 3-nitrobenzanthrone (3-NBA)40,41,42,43,44,45 and its reductive counterpart, 3-aminobenzanthrone (3-ABA),46,47,48 plant alkaloid aristolochic acid,32,33,34,35,36,37,38,39 and an aromatic amine o-anisidine.49,50,51,52 Further, the enzymes determining biological effects of these chemicals were determined employing the described methods.
Here, representative results on oxidative activation of ellipticine by P450s and peroxidases resulting in generation of covalent adducts with DNA, and on reduction of 3-NBA to metabolites that covalently modified DNA, are shown.
The plant alkaloid ellipticine (5,11-dimethyl-6H-pyrido[4,3-b]carbazole) and its derivatives are antitumor agents, which act as DNA-damaging drugs through several mechanisms, included into arrest of cell cycle and induction of apoptosis (for an overview, see29,30,31). Using the protocols described in this work, we demonstrated that the anticancer drug generates covalent DNA adducts after metabolic bioactivation catalyzed by microsomal P450s (Figure 3) and peroxidases (Figure 4)29,30,31,53,54,55,56,57,58,59,60,61, and this explained the strong efficiency of this agent against cancer cells30. The [3H]-radiolabeled ellipticine and the nuclease P1 version of the 32P-postlabeling technique were predominantly utilized in the studies29,30,31,53,61. Using the complex study with subcellular enzyme systems, the enzyme inhibitors and pure enzymes in the experiments utilizing the described protocols, the predominant P450 enzymes oxidizing ellipticine to reactive species forming DNA adducts and structures of these reactive species were characterized29,30,31,55. Of the P450s examined, the human CYP3A4 enzyme is most efficient in ellipticine oxidation to 12-hydroxyellipticine and 13-hydroxyellipticine, the ellipticine metabolites, which spontaneously decompose to ellipticine-12-ylium and ellipticine-13-ylium binding to DNA (Figure 5).55,61 The CYP enzymes also generate further metabolites such as 9-hydroxyellipticine, which is considered a detoxification metabolite, 7-hydroxyellipticine and ellipticine N2-oxide, which are formed as the minor ellipticine metabolites. The 9-hydroxyellipticine as well as 7-hydroxyellipticine and ellipticine N2-oxide are mainly formed by CYP1A1 and CYP2D6, respectively.55,57,58
Peroxidases (i.e., horseradish peroxidase (HRP), lactoperoxidase (LPO), myeloperoxidase (MPO) and cyclooxygenases (COX-1 and COX-2)) metabolize ellipticine to generate the same ellipticine-derived DNA adducts (Figure 4)61 by the mechanisms shown in Figure 5.
The nitroaromatic 3-NBA (3-nitro-7H-benzdeanthracen-7-one) is a component of diesel exhaust and is found in airborne particles62,63,64. The major metabolite of this pollutant, 3-ABA,64,65 was detected in urine of workers of salt mines that were exposed to diesel emissions for a long time63. This finding demonstrated that these workers were exposed to 3-NBA. This nitroaromatic causes lung tumors in rats after intratracheal instillation67. 3-NBA also acts as a mutagen in the Ames Salmonella typhimurium test (in strain YG1024 overexpressing nitroreductase and O-acetyltransferase), generating more than 6 million revertants per nanomole in this strain62. Its genotoxic potency was also demonstrated by generation of covalent adducts with DNA in vitro, after activation by several enzymes, using the protocols described in this work, and in vivo in several organs of rodent animals (Figure 6)39,40,41,42,43,44,45,46,47,64,67,68.
The 3-NBA-derived DNA adducts formed after 3-NBA activation with cytosolic reductases (i.e., NQO1) were measured by the nuclease P1 and n-butanol enrichment methods of the 32P-postlabeling method described in the protocols presented in this study. The results indicated the formation of up to five DNA adducts (adduct spots 1-5 in Figure 7), and three of them were characterized to be 2-(2'-deoxyadenosin-N6-yl)-3-aminobenzanthrone (dA-N6-C2-ABA; adduct spot 1), N-(2'-deoxyguanosin-N2-yl)-3-aminobenzanthrone (dG-N2-C2-ABA; adduct spot 3) and N-(2'-deoxyguanosin-8-yl)-3-aminobenzanthrone (dG-C8-N-ABA; adduct spots 4 and 5) (Figure 7 and Figure 8). Utilizing the nuclease P1 version of the 32P-postlabeling method, the dG-C8-N-ABA (adduct spots 4 and 5) was undetectable (Figure 7 and Figure 8). This result underlines that some limitations of this version of 32P-postlabeling occur, namely, the low (if any) detection of adducted deoxynucleotides that are dephosphorylated by nuclease P1 (i.e., adducts formed during oxidation of arylamines or by reduction of aromatic nitroderivatives to N-hydroxyarylamine-derivatives substituted at C8 of deoxyguanosine). Similar to the study with ellipticine, utilizing the complex study with subcellular enzyme systems, the enzyme inhibitors, pure enzymes, and DNA adduct standards in the experiments employing the protocols described in this work, the predominant cytosolic reduction enzymes metabolizing 3-NBA to metabolites generating DNA adducts, namely, the reactive metabolite formed by reduction of 3-NBA (N-OH-ABA), and structures of three DNA adducts generated by 3-NBA were characterized (Figure 7 and Figure 8). In the liver, bioactivation of 3-NBA in vitro was found to be mainly attributable to human and rat NQO1 (Figure 7), while human N,O-acetyltransferases (NATs), NAT2, NAT1, sulfotransferase (SULT), SULT1A1 and, to a lesser extent, SULT1A2 are the predominant enzymes of phase II activating 3-NBA42. Hepatic microsomal POR is also effective in the activation of 3-NBA41, but in mice, 3-NBA is mainly bioactivated by NQO1 rather than this microsomal POR42. In lung, which is the target tissue for 3-NBA carcinogenicity67, both NQO1 and XO reduce 3-NBA to metabolites generating DNA adducts. However, XO seems to act as a minor 3-NBA activating enzyme in this organ69.
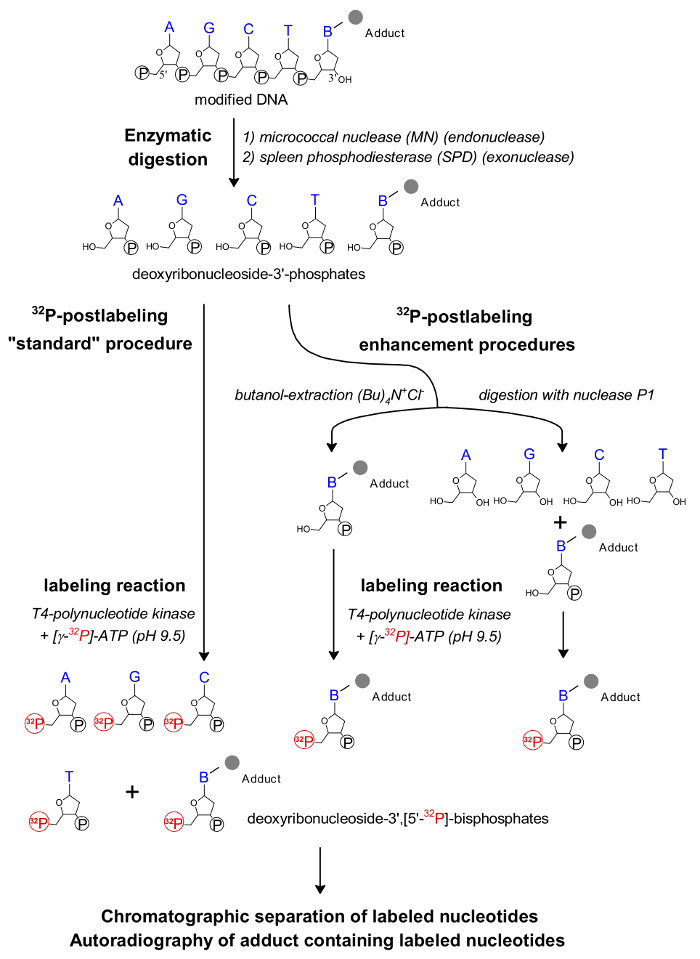
Figure 1: Scheme of the 32P-postlabeling assay. The individual steps of the 32P-postlabeling method and its enhanced procedures are shown. Please click here to view a larger version of this figure.
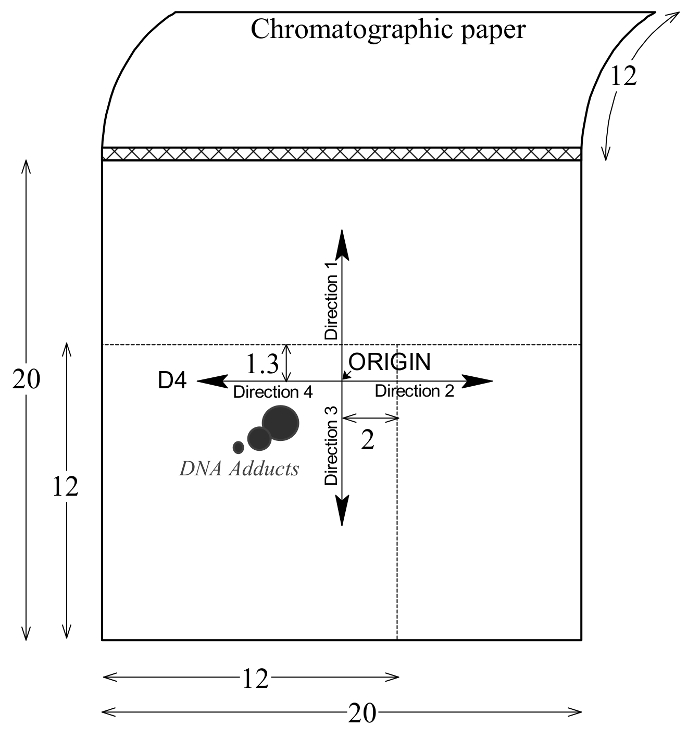
Figure 2: Pattern of DNA adduct elution on PEI-cellulose TLC plates. The multidirectional chromatography of DNA adducts on PEI-cellulose plates are shown. ORIGIN is a start position on the PEI-cellulose TLC plate. Please click here to view a larger version of this figure.
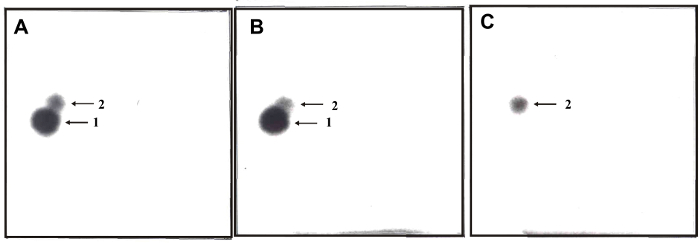
Figure 3: 32P-postlabeling analyses of DNA adducts formed in calf thymus DNA incubated with ellipticine, NADPH and (A) rat and (B) human hepatic microsomes, (C) a control sample without microsomes. Adducts 1 and 2 assigned by arrows are generated in deoxyguanosine in DNA by ellipticine activated with microsomes.32P-postlabeling was performed employing the nuclease P1 version of the method (Step 2.5.2.) Origins are located at the bottom left corners (D3 from bottom to top and D4 from left to right). D2 was omitted. Please click here to view a larger version of this figure.
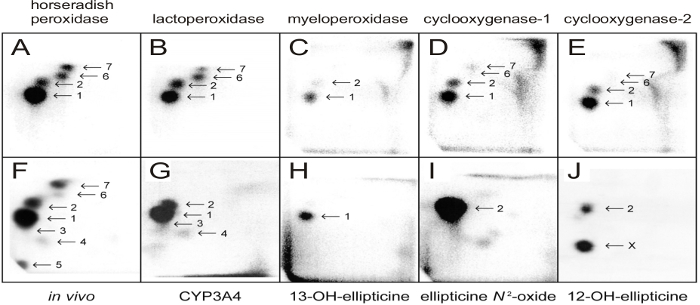
Figure 4: 32P-postlabeling analyses of ellipticine-mediated DNA adducts. Adducts formed in calf thymus DNA reacted with ellipticine (100 µM) and horseradish peroxidase (A), bovine lactoperoxidase (B), human myeloperoxidase (C), ovine cyclooxygenase-1 (D), human cyclooxygenase-2 (E) (5 µg peroxidases were present in the incubations), from liver DNA of rats treated with 40 mg ellipticine per kilogram body weight (bw) (F), from calf thymus DNA reacted with ellipticine and human CYP3A4 (G), with 13-hydroxyellipticine(H), ellipticine N2-oxide (I), and 12-hydroxyelipticine (J). Experiments were performed using the nuclease P1 version of the assay (step 2.5.2.) Origins are at the bottom left corners (D3 from bottom to top and D4 from left to right). D2 was omitted. Please click here to view a larger version of this figure.
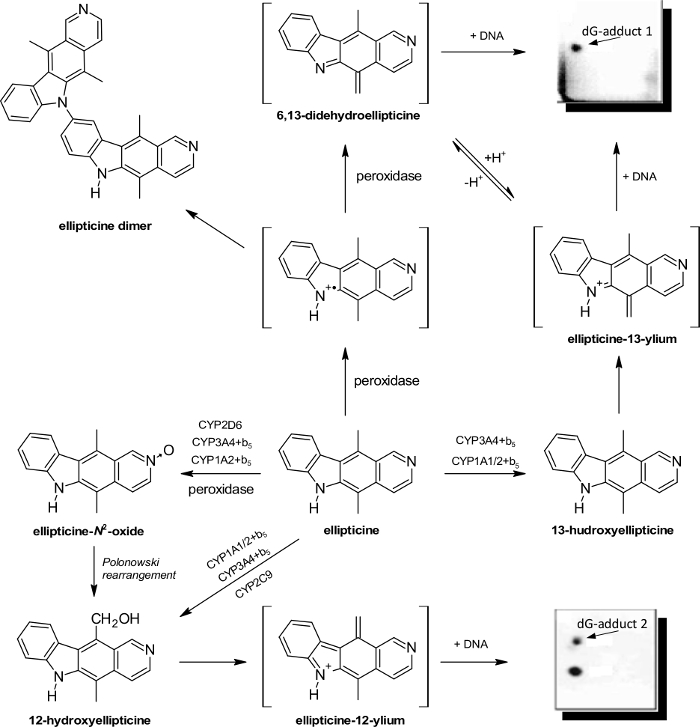
Figure 5: Oxidation of ellipticine by peroxidases and CYPs showing the ellipticine metabolites and those suggested to generate DNA adducts. The compounds in brackets have not still been detected under the experimental conditions used in the experiments, and they are the electrophilic metabolites postulated as ultimate species binding to DNA. Please click here to view a larger version of this figure.
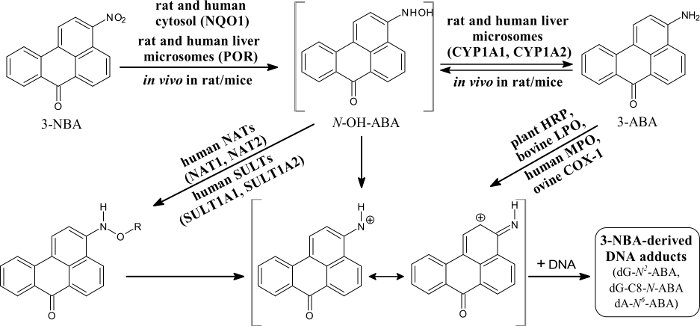
Figure 6: Metabolic activation of and DNA adduct formation by 3-nitrobenzanthrone. 3-NBA, 3-nitrobenzanthrone; NQO1, NAD(P)H:quinone oxidoreductase; NAT, N,O-acetyltransferases; SULT, sulfotransferase; CYP, cytochrome P450; POR, NADPH:cytochrome P450 oxidoreductase; HRP, horseradish peroxidase; LPO, lactoperoxidase; MPO, myeloperoxidase; COX-1, cyclooxygenase-1. R = -COCH3 or -SO3H; dA-N6-ABA, 2-(2'-deoxyadenosin-N6-yl)-3-aminobenzanthrone; dG-N2-ABA, N-(2'-deoxyguanosin-N2-yl)-3-aminobenzanthrone; dG-C8-N-ABA, N-(2'-deoxyguanosin-8-yl)-3-aminobenzanthrone. Please click here to view a larger version of this figure.
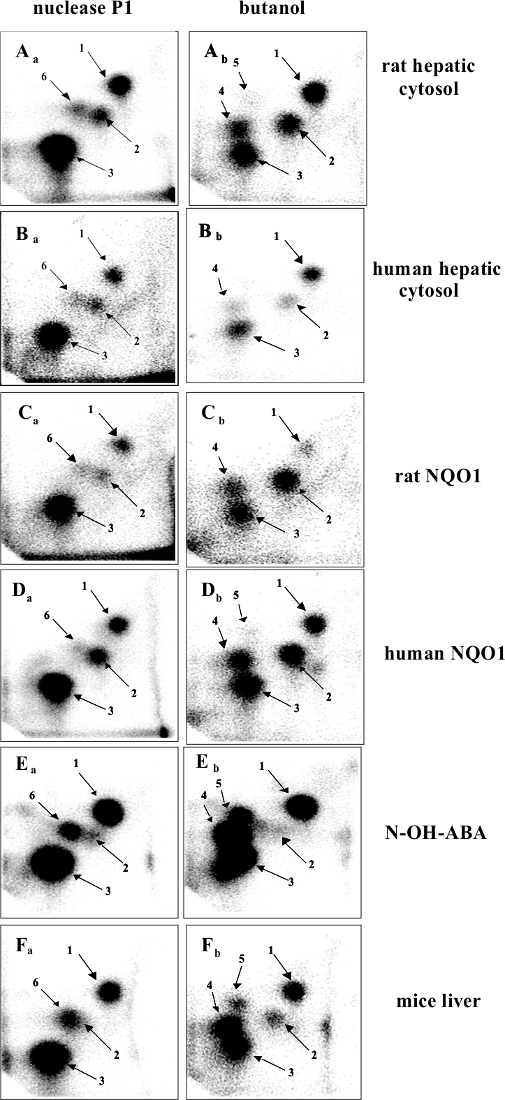
Figure 7: 32P-postlabeling analyses of 3-NBA-derived DNA adducts. The nuclease P1- (left panels) and n-butanol extraction versions (right panels) of the method were utilized. Aa and Ab, adducts formed in calf thymus DNA reacted with 3-NBA (300 µM) after activation with rat hepatic cytosols. Ba and Bb, adducts formed in calf thymus DNA, reacted with 3-NBA (300 µM) after activation with human hepatic cytosol (pooled fraction). Ca and Cb, adducts formed in calf thymus DNA, reacted with 3-NBA (300 µM) after activation with pure rat hepatic NQO1 (0.09 units). Da and Db, adducts formed in calf thymus DNA, reacted with 3-NBA (30 µM) after activation with human recombinant NQO1 (0.06 units). Ea and Eb, adducts formed in salmon testis DNA treated with N-OH-ABA. Fa and Fb, adducts formed in liver DNA of wild-type littermates on a C57BL/6 background exposed to 2 mg of 3-NBA per kg b.w. Please click here to view a larger version of this figure.
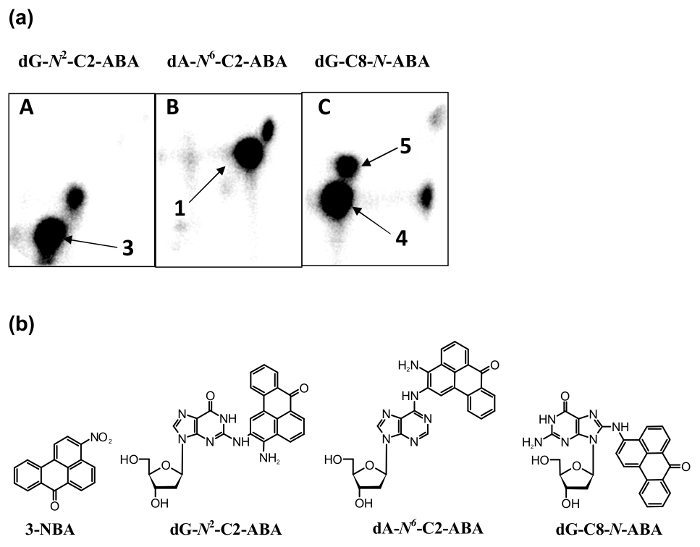
Figure 8: 32P-postlabeling analyses of 3-NBA-derived DNA adduct standards [dG-N2-C2-ABA (A), dA-N6-C2-ABA (B) and dG-C8-N-ABA (C)] (panels a) and structures of 3-NBA and these 3-NBA-DNA adducts (panels b). The n-butanol extraction version of the method was utilized (panels in a). Please click here to view a larger version of this figure.
