Note: In this study, the E14 ES cell line was used. However, this protocol is directly applicable to any other mouse ES cell line expressing a protein of interest fused to a SNAP-tag, either by tagging the endogenous protein or by using overexpression. For the examples shown in the results section, doxycycline-inducible SNAP-tag fusion cell lines were used (SNAP-tag fused to the following proteins: Nanog, Oct4, Srsf11, or to the fluorescent proteins mOrange2 and sfGFP, and put under the control of a doxycycline-inducible promoter. See11 for further information on the plasmids used for the generation of the doxycycline-inducible SNAP-tag fusion cell lines). The doxycycline-inducible system can be particularly useful, as it allows for tightly controlling the timing and intensity of the expression of the protein of interest. C-terminal positioning of the SNAP-tag is recommended, as changing the N-terminal amino acid sequence is more likely to alter the half-life of the target protein (N-end rule12).
1. E-cadherin coating and cell seeding
- Generate an ES cell line expressing a protein of interest fused to the SNAP-tag4.
- Coat 100 mm cell culture dishes with 4 mL of 0.1% gelatin (diluted in phosphate buffered saline (PBS) without Ca2+/Mg2+) for 1 h. Remove gelatin and let dry for 1 h. Use immediately or store the coated dishes for up to 2 months.
- Grow the cell line of interest in ES cell culture medium (Glasgow Minimum Essential Medium, supplemented with 10% ES cell-qualified fetal bovine serum, 2 mM sodium pyruvate, 1% non-essential amino acids, 1% penicillin/streptomycin, 2 mM L-glutamine, 100 µM 2-mercaptoethanol, leukemia inhibitory factor (LIF), 3 µM CHIR99021 and 0.8 µM PD184352) on gelatin-coated dishes at 37 °C and 5% CO2 for 2 days until reaching a confluence of 10 – 20 Mio cells per dish.
Note: Here, LIF was produced by transient transfection of HEK 293T cells, followed by supernatant collection and filtering. Each batch of LIF was tested for its potential to maintain pluripotency, but concentrations were not determined. However, recombinant LIF is also commercially available and the concentration commonly used for mouse ES cell culture is 1000 units/mL13. - Coat a 96-well plate suitable for imaging with 30 µL of recombinant mouse E-cadherin Fc chimera protein (E-cad-Fc) per well (5 ng/µL, diluted in PBS with Ca2+/Mg2+. Use stock concentrations of 100 ng/µL, stored at -80 °C). Avoid extensive pipetting, as E-cad-Fc is very fragile. Incubate at 37 °C for 1.5 h.
Note: Depending on the microscope, other formats might be used. - Aspirate the E-cad-Fc, wash once with 100 µL of PBS with Ca2+/Mg2+, and add 100 µL of pre-warmed ES cell culture medium.
- Wash the cells of interest with 5 mL of PBS without Ca2+/Mg2+. Aspirate and add 2 mL of Trypsin-EDTA 0.25%. Incubate for 4 min at 37 °C. Add 4 mL of ES cell culture medium, resuspend and spin down at 1000 x g for 4 min. Aspirate the supernatant and resuspend the pellet in 2 mL of fresh ES cell culture medium.
Note: PBS without Ca2+/Mg2+ should be used for washing the cells before trypsinization in order to remove Ca2+ ions, which are required for cell adhesion. In contrast, for the E-cad-Fc dilution and coating (step 1.4), use PBS with Ca2+/Mg2+, since the adhesion of ES cells to E-cad-Fc coated dishes is Ca2+-dependent14. - Dilute the resuspended cells 1:10 in 1 mL of ES cell culture medium and count, using a counting chamber (load 10 µL for a chamber of 0.1 mm depth).
- Seed 30,000 cells/cm2 in the E-cad-Fc coated imaging plate. Fill up to 200 µL per well with ES cell culture medium. For doxycycline-inducible cell lines, induce with an appropriate dose of doxycycline (optimize dose and timing of the induction in advance. The cell lines used here were treated with 500 ng/mL doxycycline 24 h before imaging). Incubate at 37 °C and 5% CO2 and proceed with the pulse labeling and imaging 24 h after cell seeding.
2. Pulse labeling of the SNAP-tag
Note: For protein decay experiments it is crucial to use an adequate SNAP dye concentration. The concentration should be high enough to yield a bright signal in the beginning of the time-lapse, as the fluorescence will decrease over time. However, using too high dye concentrations might cause residual dye being left in the medium or in the cells even after washing. The free dye might subsequently bind to newly produced SNAP-tag molecules over the course of the movie, which will distort the decay curve. The observed fluorescence signal will depend on the properties of the dye, the cell line used, as well as the expression level of the corresponding protein. Therefore, it is crucial to optimize the dye concentration by testing different dilutions, starting from the dilution suggested for live cell imaging by the manufacturer. For this study, a far-red fluorescent substrate was used. An optimal concentration of 12 nM was determined for doxycycline-inducible overexpression cell lines.
- Dilute the SNAP dye to the appropriate concentration in pre-warmed ES cell culture medium. Use a pipette to gently remove the medium from the cells previously seeded in the 96-well plate and add 50 µL of diluted SNAP dye per well. Incubate for 30 min at 37 °C.
- Aspirate the diluted dye with a pipette and wash 3x with 200 µL of pre-warmed PBS (without Ca2+/Mg2+). Add 200 µL of ES cell culture medium and incubate for 15 min at 37 °C.
- Repeat the previous washing steps twice more.
Note: Extensive washing is important in order to remove any unbound dye. The repeated washing steps with PBS remove the residual extracellular dye in the medium, whereas the 15 min incubations ensure robust labeling of the SNAP-tag fusion proteins prior to starting the imaging experiment. However, perform the washing steps by gentle pipetting and do not use an automatic aspirator, as cells seeded on E-cad-Fc tend to detach easily. - Add 200 µL of imaging medium. To reduce the fluorescence background, use phenol red free medium (supplemented with 10% ES cell-qualified fetal bovine serum, 2 mM sodium pyruvate, 1% non-essential amino acids, 1% penicillin/streptomycin, 2 mM L-glutamine, 100 µM 2-mercaptoethanol, LIF, 3 µM CHIR99021 and 0.8 µM PD184352).
3. Time-lapse microscopy
- Use a microscope suitable for live imaging, allowing controlled temperature and CO2. Set the temperature to 37 °C and the CO2 to 5% prior to use and allow to equilibrate for 1 – 2 h.
- Place the plate into the microscope and find spots suitable for imaging. Select spots where the cells are evenly distributed, but not too dense, as this will facilitate the analysis. Adjust the number of selected spots to the number of cells required for the analysis. In this study, 3 - 5 spots per cell line were recorded.
- Select the objective. A 20X objective is appropriate for the size of ES cells.
- Select the illumination settings for the fluorescent channel(s) to be recorded. Adjust the laser power and exposure according to the intensity of the fluorescence signal from the cells of interest. Make sure to obtain a strong initial signal, since it will decrease over the course of the movie, but avoid laser powers higher than 30% in order to minimize phototoxicity and photobleaching. For this study, an excitation filter of 632/22 nm (wavelength/bandpass), emission filter of 679/34 nm (wavelength/bandpass), laser power of 10% and exposure time of 100 ms were used.
- Select the imaging intervals and duration of the time-lapse experiment. For proteins with expected half-lives of 2 – 20 h, choose acquisition times of 12 – 24 h with time intervals of 15 min. For shorter-lived proteins, reduce the intervals and acquisition times, for longer-lived proteins increase accordingly.
- Start imaging.
4. Image processing and analysis
- Collect the acquired images as a stack in tiff format. For further processing and analysis, use the FIJI software15. If the time-lapse data is not collected as a stack by the microscope software, open all frames of the time-lapse experiment in the software (either by clicking File | Open… or drag and dropping the corresponding files to the toolbar) and click Image | Stacks | Images to Stack.
- Use the subtract background function to remove the background from all pictures in the stack. To do so, click Process | Subtract Background. Select the rolling ball radius in the pop-up window. Use a radius that is at least the size of the cells imaged. To apply the background subtraction to all cells in the stack, select Yes in the Process stack? pop-up window.
- Select a cell of interest and draw a region of interest (ROI) around it using the toolbar. For nuclear signals, use the Oval selection by first clicking on the corresponding tab in the toolbar and then clicking on the nucleus of interest and adjusting the oval around it. For cytoplasmic signals or very dense regions use the freehand selection to be able to closely follow the cell outlines. Avoid including too large regions of background, even though it is not necessary to follow the cell outlines precisely due to the subsequent subtraction of all background pixels (step 4.8 – 4.9).
- Add the region of interest to the ROI manager by clicking Analyze | Tools | ROI manager | Add, or by pressing T on the keyboard.
- Proceed to the next frame by moving the tab in the stack window or by pressing shift + < on the keyboard, then repeat the previous actions. Follow the cell of interest throughout the course of the movie and add the ROI of each frame to the ROI manager. Save the final ROI set for each tracked cell by clicking Daha | Save… in the ROI manager.
Note: After cell divisions, track both daughter cells separately and sum their integrated intensities after background subtraction (see steps 4.6 – 4.9) to account for protein dilution during cell division. Save the ROI sets for both daughter cells. - Once the cell of interest is tracked throughout the movie, click Measure, to obtain values for the mean intensity and the area of the ROIs. Copy the obtained values into an electronic spreadsheet program and calculate the raw integrated intensity of the cell for each time-point as follows:
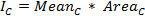
where Meanc is the mean intensity and AreaC the area of the corresponding ROI. - To estimate the local background, add a ROI close to the cell of interest for each time frame. Avoid including any cellular fluorescence signal. Use a circular ROI that is roughly the size of a cell and move or shrink it accordingly if needed, e.g. if neighboring cells interfere. Proceed as described previously with the cellular ROI set in order to obtain a background ROI set and copy the measured intensity values to the spreadsheet.
- To obtain a background-corrected value for the integrated intensity of the cell, first calculate the integrated intensity of the background for each time-point:
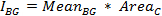
where MeanBG is the mean intensity of the background signal and AreaC is the area of the ROI encircling the cell. Do not use the area of the background ROI, unless both areas have the same size. - Calculate the final background-subtracted integrated intensity of the cell for each time point:

- To normalize the single cell decay curves, divide the intensity value of each time-point by the intensity value of the first time-point.
Note: The curve fitting (see step 4.11) can either be performed on each single cell or on the population average. A normalization is required if a population based average is calculated in order to avoid biases from different fluorescence intensities between cells. The normalization thus ensures that each cell contributes with the same weight to the final decay curve. In addition, the normalization can be useful to visualize the single cell decays independently of their absolute fluorescence intensities (see Figures 3A-3C). However, if only the single-cell half-lives are determined and the data is not averaged, the normalization step may be omitted and the curve fitting can be directly performed on the raw data. - For the estimation of the protein half-life, use a curve fitting tool. In this study, the MATLAB curve fitting toolbox 3.4.1 was used, which is located by default in the Apps section of the MATLAB user interface. Import the fluorescence intensity and time values from the electronic spreadsheet into MATLAB by clicking on the Import Data tab. Open the curve fitting toolbox and select the time points and fluorescence decay data in the X data and Y data tab. Choose Custom Equation in the curve fitting tab and enter the equation for an exponential decay:
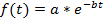
where f(t) is the fluorescence intensity at a given time-point, a the initial intensity and b the decay rate. In the Fit Options… tab, select 0 for the lower limit of both a and b. The estimated values for a and b will then appear in the results window. Calculate the half-life as follows:
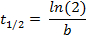
The described protocol provides an estimate of the cell-to-cell variability in half-life for any given protein fused to a SNAP-tag. The use of recombinant E-cadherin-Fc for coating of the imaging plate allows for single cell resolution in ES cells, which otherwise grow in colonies. Single cells can be tracked separately throughout the course of the movie ( Figure 1A).
In order to determine the protein half-life for each single cell, it is crucial to measure the integrated, background-subtracted SNAP-tag fluorescence signal over time ( Figure 1B), with summing up the integrated intensities of both daughter cells in case of divisions. This results in an exponential decay curve for each cell, from which the decay rate and thus the half-life can be extracted by curve fitting ( Figure 1C). Importantly, if an average decay curve is calculated, the single cell traces should be normalized to the first frame to ensure that each cell has the same weight on the average, despite putative differences in initial fluorescence intensity between cells.
The adequate dye concentration depends on the type of dye and cell line used, and should thus be optimized for each experiment. To illustrate this, Figure 2 shows the decay curves of different concentrations of the far-red fluorescent substrate used for all experiments shown here, tested on a doxycycline inducible SNAP-tag fusion cell line. An initial dye concentration of 3 µM was chosen according to the manufacturer's instructions and a serial dilution of 1:3 was performed until reaching a concentration of 1.4 nM. For the two highest concentrations the signal does not decrease, whereas the observed decay is exponential for concentrations below 111 nM. An optimal concentration of 12 nM of dye was chosen for further experiments, since it ensured minimal amounts of residual dye left in the medium after washing, but the signal was still bright enough to observe clear decay curves for a variety of doxycycline inducible cell lines.
Figures 3A-3C show representative results, including the single cell decays and population averages, for 3 proteins with different average half-lives: the pluripotency associated transcription factors Nanog (average half-life 2.9 h) and Oct4 (average half-life: 4.8 h), and the much longer lived pre-mRNA processing factor Srsf11 (average half-life 25.3 h). The boxplots ( Figure 3D) represent the half-lives obtained from the single cell decay curves by curve fitting and illustrate the heterogeneity of half-lives for the three proteins.
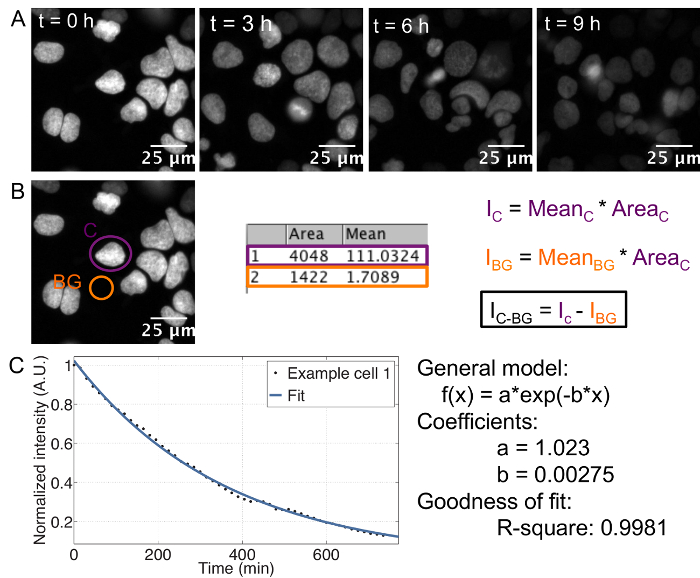
Figure 1: Time-lapse acquisition and analysis
A: Series of representative images showing the decay of fluorescence in cells pulse labeled with SNAP dye. Cell line: doxycycline inducible TRE3G-Oct4-SNAPtag, induced with 500 ng/mL doxycycline 7 h before imaging.
B: Movie analysis and calculation of integrated intensity. Ic: Integrated intensity of the cell, IBG: Integrated intensity of the background, IC-BG: background-corrected integrated intensity.
C: Exponential curve fitting performed in MATLAB. Example of a single cell decay trace, the corresponding fit and the resulting estimates for the decay parameters a and b. The half-life is calculated as follows: t1/2 = ln(2)/b. Please click here to view a larger version of this figure.
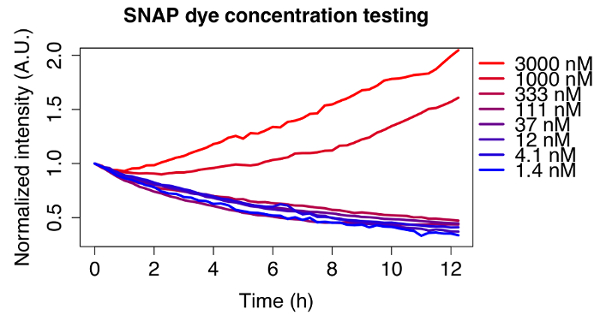
Figure 2: Optimization of the dye concentration.
Fluorescence decay of cells pulse labeled with different concentrations of SNAP dye. A serial dilution of the dye starting from 3 µM was performed. The traces are averages of 5 cells each, normalized to the first frame and tracked for 12 h. Cell line: doxycycline inducible TRE3G-sfGFP-mOrange2-SNAPtag-PEST, induced with 500 ng/mL doxycycline 24 h before imaging. Please click here to view a larger version of this figure.
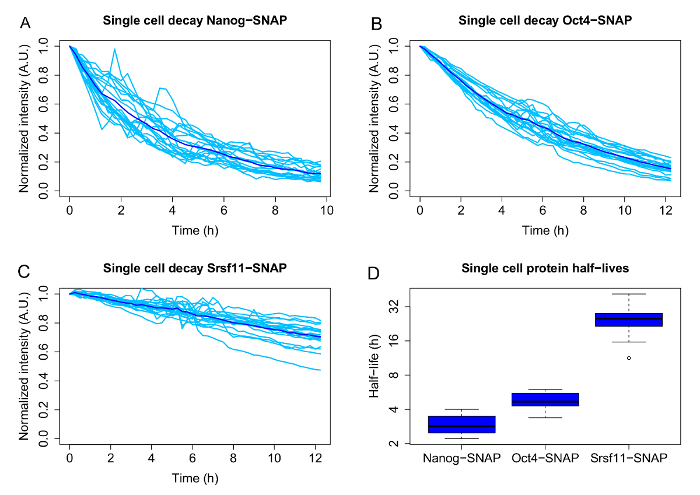
Figure 3: Single cell variability of half-lives for different SNAP-tagged candidate proteins.
A-C: Single-cell decays (light blue) and population averages (dark blue) for doxycycline inducible Nanog-, Oct4- and Srsf11-SNAP-tag fusion proteins.
D: Half-lives of individual cells, which were calculated by exponential curve fitting for each single cell. The boxplots are displayed in log2 scale. The boxes display the interquartile range (IQR), the black line indicates the median. The whiskers represent the minimum and maximum values, excluding the outliers (black dots), which are defined as values deviating more than 1.5x from the IQR. N = 20 cells each. Please click here to view a larger version of this figure.
