Expansion and Adipogenesis Induction of Adipocyte Progenitors from Perivascular Adipose Tissue Isolated by Magnetic Activated Cell Sorting
Özet
Here we report a method for isolation of Adipocyte Progenitor Cell (APC) populations from Perivascular Adipose Tissue (PVAT) using Magnetic-activated Cell Sorting (MCS). This method allows for an increased isolation of APC per gram of adipose tissue when compared to Fluorescence-Activated Cell Sorting (FACS).
Abstract
Expansion of Perivascular Adipose Tissue (PVAT), a major regulator of vascular function through paracrine signaling, is directly related to the development of hypertension during obesity. The extent of hypertrophy and hyperplasia depends on depot location, sex, and the type of Adipocyte Progenitor Cell (APC) phenotypes present. Techniques used for APC and preadipocytes isolation in the last 10 years have drastically improved the accuracy at which individual cells can be identified based on specific cell surface markers. However, isolation of APC and adipocytes can be a challenge due to the fragility of the cell, especially if the intact cell must be retained for cell culture applications.
Magnetic-activated Cell Sorting (MCS) provides a method of isolating greater number of viable APC per weight unit of adipose tissue. APC harvested by MCS can be used for in vitro protocols to expand preadipocytes and differentiate them into adipocytes through use of growth factor cocktails allowing for analysis of the prolific and adipogenic potential retained by the cells. This experiment focused on the aortic and mesenteric PVAT depots, which play key roles in the development of cardiovascular disease during expansion. These protocols describe methods to isolate, expand, and differentiate a defined population of APC. This MCS protocol allows isolation to be used in any experiment where cell sorting is needed with minimal equipment or training. These techniques can aid further experiments to determine the functionality of specific cell populations based on the presence of cell surface markers.
Introduction
Perivascular Adipose Tissue (PVAT), due to its close proximity to blood vessels, is a major paracrine signaling component in vasculature function1. Expansion of this adipose tissue is dependent on the phenotype of the Adipocyte Progenitor Cells (APC) present2,3. Isolation of cells from adipose tissues is difficult as primary adipocytes are fragile, buoyant, and range in size. Certain isolation techniques can also alter cell phenotype and morphology by increasing inflammatory protein synthesis and reducing adipogenic gene expression4, emphasizing the importance of a protocol that maintains the integrity of the cells.
Culture of primary cells and specific preadipocyte subpopulations gives a reductionist approach to in vivo growth and maintains equivalent cellular genetic makeup5, although working time with these cells is limited due to deterioration with aging, or senescence6. Preadipocytes from various adipose depots, including subcutaneous and omental depots, also demonstrate differences in proliferation7, which emphasizes the importance of collecting cells from specific anatomical sites. Precursor cells from non-PVAT white adipose depots have been characterized in previous studies7,8,9, but less is known about PVAT APC phenotypes.
The techniques described here allow for the analysis of specific and defined APC populations with minimal impact on their morphology, viability, and potential to proliferate and differentiate. Magnetic-activated Cell Sorting (MCS) is amenable to downstream applications, such as culture, as the beads dissolve without altering the cell. MCS is also economical, and once the antibody concentrations have been standardized, the need for flow cytometry assays is minimal. In vitro studies with PVAT precursors can also give a glimpse of the potential that these primary cells may have.
Protocol
All procedures described in this paper follow guidelines established by the Institutional Animal Care and Use Committee (IACUC) of Michigan State University. All buffers and medias should be protected from light.
1. Preparation of Buffers, Media, and Instruments
- Prepare Krebs Ringer Bicarbonate-Buffered solution (KRBB): 135 mM sodium chloride, 5 mM potassium chloride, 1 mM magnesium sulfate, 0.4 mM potassium phosphate dibasic, 5.5 mM glucose, 1% antibiotic/antimycotic (10,000 units/mL penicillin, 10,000 µg/mL streptomycin, 25 µg/mL amphotericin B), and 10 mM HEPES (pH = 7.4). This solution is stable for 3 weeks when kept sterile and at 4 °C.
- Prepare Collagenase Type 1 Solution: 1 mg/mL in KRBB with 4% Bovine Serum Albumin (BSA). This solution should be kept at 37 °C and is stable for 4 h.
- Prepare Erythrocyte Lysis Buffer Solution: 154 mM ammonium chloride, 10 mM potassium bicarbonate, and 0.1 mM EDTA. Keep at 4 °C for up to one month.
- Prepare MCS Blocking Buffer: DMEM/F12 Media base, 10% Fetal Bovine Serum (FBS), 5% normal donkey serum, 40 µL/mL F(ab) Fragment Donkey Anti-Rat IgG. Keep at 4 °C for up to one month.
- Prepare MCS Buffer Solution: Phosphate-Buffered Saline (PBS, pH = 7.5), 0.5% BSA, and 2 mM EDTA. Keep at 4 °C for up to one month. De-gas solution by heating to 37 °C in a glass container and then applying a vacuum for15 s. Leave unused buffered sealed.
- Prepare Stromal Vascular Fraction (SVF) Basal Media: Dulbecco's-Modified Eagles Medium (DMEM):F12, 15% Fetal Calf Serum (FCS), 1% antibiotic/antimycotic (10,000 units/mL penicillin, 10,000 µg/mL streptomycin, 25 µg/mL amphotericin B), 44.05 mM sodium bicarbonate, 100 µM ascorbic acid, 33 µM biotin, 17 µM pantothenate, 2 mM L-glutamine, and 20 mM HEPES. Keep sterile and at 4 °C for up to 2 weeks.
- Prepare APC Media: Basal Media with additional growth factors including epidermal growth factor 10 ng/mL), leukemia inhibitory factor (10 ng/mL), platelet-derived growth factor BB (10 ng/mL), and basic fibroblast growth factor (5 ng/mL). Keep sterile and at 4 °C for up to 2 weeks.
- Prepare APC Induction Media: APC Media with 10% FBS, 2.5 µg/mL insulin, 0.5 mM 2-isobutyl-1-methylaxanthine (IBMX), 1 µM dexamethasone, and 200 pM T3 (triiodothyronine thyroid hormone). Keep sterile and at 4 °C for up to 2 weeks.
2. Adipocyte Progenitors Isolation
- Anesthetize rat according to institutional guidelines. Place the rat in dorsal recumbency. Confirm depth of anesthesia via a toe-pinch and the loss of reflex response to this painful stimulus.
NOTE: This protocol uses 10-week-old Sprague Dawley rats and 70 mg/kg of pentobarbital delivered via an intraperitoneal injection. - Make a vertical midline incision with scissors along the sternum in the thoracic area and to the perineal area. Access the abdominal cavity and expose the superior mesenteric artery, the small mesenteric resistance vessels (mPVAT) and the thoracic aorta (aPVAT).
- Sever all connections to the mesentery and aorta and remove vessels from animal. Isolate PVAT by using a dissecting microscope and Petri dish filled with KRBB to view vessels and isolate PVAT.
- In this experiment, collect gonadal (GON) adipose to represent a non-PVAT adipose depot. Place isolated fat pads on ice in KRBB with 10 mM HEPES (pH = 7.4).
- In a biosafety hood, transfer about 50 mg of tissue to a 1.7 mL tube with 1 mL of collagenase type I solution and mince with tissue scissors (1 – 3 mm pieces).
- Digest samples by incubating at 37 °C in a rotisserie incubator (or incubator with an orbital shaker) for 1 h. In a biosafety hood, sequentially filter digested material through 100 and 40 µm cell strainers into a 50-mL tube. Centrifuge resulting filtrate at 4 °C for 10 min at 300 × g.
NOTE: All steps in the protocol from here forward are to be performed in a biosafety hood to keep cells sterile. - Pour off supernatant and resuspend pellets containing the SVF cells in 1 mL of 1X Erythrocyte Lysis Buffer Solution and transfer to a 1.7 mL microfuge tube. Incubate cells for 5 min at RT protected from light and centrifuge at 4 °C for 5 min at 300 x g.
- Pour off supernatant and resuspend remaining cell pellet in SVF Basal Media. Collect a 20 µL sub-sample to count live cells with Trypan Blue Solution.
NOTE: The number of SVF cells that can be isolated will vary by site. Average numbers of SVF harvested per mg of tissue are: aPVAT = 5.0 ±2.0×103, mPVAT = 1.04 ±0.62×104, GON = 2.4 ±1.2×105.
3. Magnetic-activated Cell Sorting
NOTE: Isolate APC from SVF based on CD34 and PDGFRα cell surface markers by performing all steps at 4 °C.
- Spin cells for 5 min at 300 x g. Pour off supernatant and resuspend the cell pellet in MCS Blocking Buffer at 1 x 106 cells/mL and incubate for 20 min.
NOTE: Cell suspensions of 1 x 106 – 2 x 108 cells/mL can be separated effectively. - Incubate cells with 5 µL of FITC-conjugated Mouse anti-CD34 (1 µg/1 x 106 cells) for 30 minutes at 4 °C. Spin cells for 5 min at 300 x g and 4 °C.
- Incubate with 4 µL of anti-FITC microbeads and 96 µL of MCS buffer (total volume of 100 µL) for 5 min at 4 °C in the dark to separate CD34+ and CD34– cells.
- Attach magnetic separator to the stand and place the MultiSort (MS) Column, with the column wings to the front, into the separator. Place a 5 mL collection tube under the MS Column in the upper tube holder.
- Prepare MS Column by rinsing with MCS Buffer Solution. Apply 500 µL of MCS Buffer on top of the column and let the buffer run through. Discard effluent and change collection tube.
- Load antibody labeled cell suspension onto the prepared MS Column. Collect flow-through containing unlabeled cells.
- Wash MS Column with 500 µL of degassed MCS Buffer 3x. Collect unlabeled cells that pass through and combine with the flow through from previous step.
- Remove MS Column from the magnetic separator and place it on a new collection tube. Pipette 1 mL of MCS Buffer onto the MS Column. Immediately flush out fraction with the magnetically labeled cells by firmly, but slowly, applying the plunger supplied with the column as to not allow excess gas into the column.
NOTE: Isolated cell numbers will vary by site. Average numbers of CD34+ APC isolated from the SVF population per mg of tissue are: aPVAT = 2.6 ±0.43×102, mPVAT = 9.6 ±1.4×102, GON = 1.3 ±0.22×103. - Spin cells for 5 min at 300 × g and 4 °C. Incubate the CD34+ fraction collected in 10 µL of a 1:200 solution/1 x 106 cells Rabbit anti-PDGFRα for 30 min at 4 °C.
- Centrifuge cells again for 5 min at 300 x g and 4 °C. Incubate labeled cells with 4 µL of anti-rabbit IgG microbeads and 96 µL of MCS buffer by repeating steps 3.3 through 3.7 to isolate.
NOTE: Isolated cell numbers will vary by site. Average numbers of PDGFRα+ APC isolated from the CD34+ population per mg of tissue are: aPVAT = 2.4 ±0.64, mPVAT = 8.4 ±2.4, GON = 10.4 ±1.9, which is 0.5 – 10% of the previously isolated population.
- Centrifuge cells again for 5 min at 300 x g and 4 °C. Incubate labeled cells with 4 µL of anti-rabbit IgG microbeads and 96 µL of MCS buffer by repeating steps 3.3 through 3.7 to isolate.
4. Cell Culture and Adipogenesis Induction
- Culture the SVF and APC in 6-Well tissue culture plates in basal media with replacement every 2 days. After 3 serial passages, plate in black 96-Well tissue culture plates at 1×102 cells/well for proliferation assays, which are evaluated at 8, 24, 48, and 96 h, and at 50,000 cells/well in 24-well plates or 10,000 cells/well in 48-Well tissue culture plates for adipogenesis assays, which are qualitative and quantitative.
- Supplement APC basal media for 48 h post-confluency and prior to induction with bone morphogenic protein 4 (3.3 nmol/L) as indicated10 for differentiation. Induce cells after 48 h of 100% confluency (day 0) using the APC Induction Media to incubate the cells.
- After 48 h, change media to maintain cells in APC Induction media without IBMX and dexamethasone, for 14 days with media changes every 48 h.
- MCS isolation validated by FACS.
- Take a 50 µL (50,000 cell) sub sample of magnetically separated cells and wash with FACS solution.
- Centrifuge cells and resuspend in 100 µL of a 1:1,000 solution of donkey anti-rabbit IgG Dylight 405 to label the PDGFRα+ cells. Incubate for 30 min protected from light and at 4 °C.
- Wash cells and resuspend in 200 µL of 2% formaldehyde solution until time for analysis using 488 nm (FITC) and 405 nm (Dylight 405) filters on a flow cytometer.
NOTE: Lipid accumulation by cultured cells is assessed quantitatively using a lipophilic adipogenesis fluorescence assay in a microplate reader measuring fluorescence and using preadipocytes as calibrators for lipid accumulation. Lipid accumulation is also measured qualitatively by lipid dye staining and imaging performed on an inverted microscope equipped with a camera, making the percentage of total cells that do or do not contain lipid observable. Any plate reader that is able to measure fluorescence with excitation at 485 nm and emission at 572 nm is suitable for analysis as well as any microscope with a camera capable of capturing digital images.
Representative Results
Proliferative capacity of preadipocytes and adipogenic potential of adipocyte precursors are characteristics that are maintained in vitro11. In vitro proliferation of isolated SVF and APC from aPVAT, mPVAT, and GON of male rats was evaluated at 8, 24, 48, and 96 h after plating using a quantitative DNA assay. No site differences in SVF expansion rate were observed at any time point except for the APC from aPVAT, which had less proliferation by 96 h compared to SVF cells from the same site (Figure 1).
Confluent APC stimulated with Bone Morphogenic Protein 4 (BMP4) for 48 h prior to standard induction12 exhibited differentiation. This was evident by greater lipid accumulation in droplets as evaluated by both fluorescent lipid uptake assay (Figure 2A) and Oil Red O staining (Figure 2B).
When comparing the yield of APC, MCS isolation produced a greater number of cells ready for culture compared to FACS. (Figure 3) Importantly, the distribution and viability of APC populations (CD34+ and PDGFRα+) was similar between MCS and FACS. Cell viability determined by Trypan Blue staining for counting post isolation was similar for both isolation procedures (FACS = 71.57% ±11.09; MCS = 79.25% ±7.47). These data demonstrate that the MCS isolation of APC yields a higher number of viable APC compared to MCS.
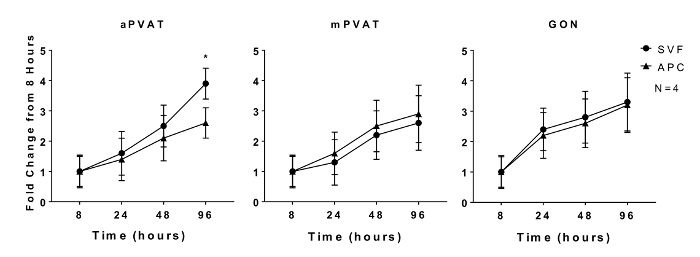
Figure 1: In Vitro Proliferation of Adipocyte Progenitor Cells (APC) is Affected by Anatomical Location. Stromal Vascular Fraction (SVF) and APC were isolated from aortic and mesenteric perivascular adipose tissue (aPVAT & mPVAT, respectively) and gonadal adipose from 10 week old male rats. Proliferation of cells was measured by a DNA quantification assay at 8, 24, 48, and 96 h time points after seeding. Data are expressed as fold increase over 8 h baseline ±SEM (N = 4). Significance is indicated by * (P <0.05). Figure modified from Contreras et al. 201613. Please click here to view a larger version of this figure.
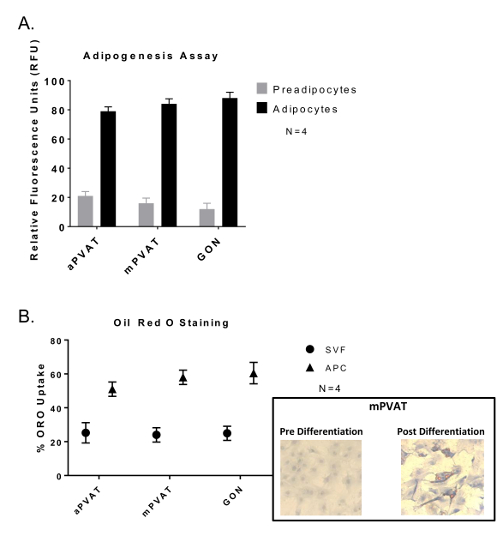
Figure 2: Adipocyte Progenitor Cells (APC) Show no Variation in Differentiation Abilities between Depots but Greater Lipid Accumulation Compared to Stromal Vascular Fraction (SVF). Cells were induced after 48 h of confluency and exposure to BMP4 followed by 48 h of exposure to dexamethasone and 3-isobutyl-1-methylxanthine and then sustained in maintenance media 14 days with media changes every 48 h. (A) Lipid uptake assay of differentiated APC with data expressed as ratio of undifferentiated preadipocytes: differentiated adipocytes (preadipocytes:adipocytes) in relative fluorescence units (RFU) ±SEM (N = 4). (B) Oil Red O (ORO) staining of APC and SVF with data expressed as percent of cells with lipid (ORO Uptake) ±SEM (N =4 ). Significance is indicated by * (P <0.05). Figure modified from Contreras et al. 201613. Please click here to view a larger version of this figure.
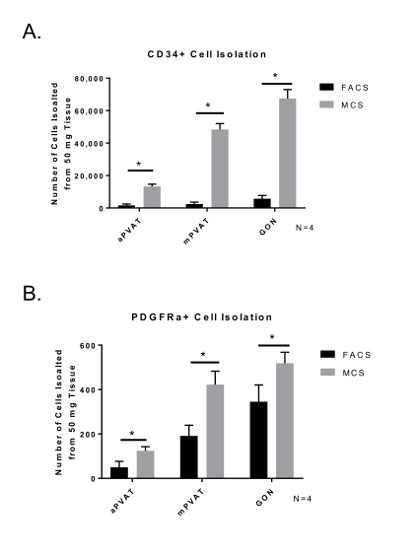
Figure 3: Isolation Yield of Viable Adipocyte Progenitor Cells (APC) is Improved by Magnetic-activated Cell Sorting (MCS). Surface marker expression of CD34+ (A) and PDGFRα+ (B) in SVF isolated from perivascular adipose tissues (aortic = aPVAT; mesenteric = mPVAT) using MCS and FACS. Data are expressed as mean number of cells isolated from 50 mg of tissue ±SEM (N = 4). Significance is indicated by * (P <0.05). Please click here to view a larger version of this figure.
Discussion
The central focus of the present experiment is the isolation, expansion, and adipogenic induction of APC from PVAT depots. Here we present a protocol for the isolation of APC based on the identification of cells expressing the surface markers CD34 and PDGFRα. These surface proteins were previously identified on APC with high proliferation rates and the potential to differentiate into white or brown adipocytes in various adipose depots14,15. By selecting cells based on these specific markers, we were able to isolate similar APC populations from multiple adipose depots that match the adipocyte phenotype that is observed in the PVAT selected13. In our experiments we improved APC differentiation by supplementing the growth factor BMP4. Previously, Macotela and colleagues demonstrated that specific APC populations from visceral adipose depots had less BMP activity than those from subcutaneous depots when differentiated unless culture media was supplemented with the adipogenic growth factors BMP2 or BMP412,16,17,18.
Since isolation of individual cells from adipose tissue is difficult due to the fragility of the cells, using MCS provides an efficient method for cell isolation at a smaller cost of consumables, equipment, and training resources. It is important to note that APC yield can be affected by the animal's age or size and by changes in the temperature of media and failure to maintain a sterile isolation environment. Culturing cells in normoxia is also important for proliferation and differentiation19, thus maintenance of incubation air conditions is integral to culture practice. Centrifugation speeds can be increased to 800 x g if sufficient cell pellets do not form during the isolation process. Checking the expiration of the antibodies may also be necessary as the conjugated FITC fluorochrome can degrade over time. Changing lots of Collagenase Type I may also require alterations to digestion procedures as lots can vary in potency. If using a different species other than rat, specific serum and IgG in the blocking buffer may also be necessary if the host species of the antibodies is different from the ones used here.
Among the advantages of MCS over FACS are that it is more economically feasible than FACS as kits are inexpensive and can be easily purchased. This also avoids the purchase, maintenance, and usage training of a flow cytometer. Using MCS also allows for specific binding without adjustment of gates for selection and background fluorescence involvement.
A limitation to this study is that it relied on two preadipocyte surface markers. Other preadipocyte surface markers of commitment and differentiation, such as Zfp423, Sca1, and CD24, have been identified20,21. These markers may accurately identify committed adipocyte progenitor cells of specific phenotypes; however, the surface markers used here were selected since cells expressing these markers have the ability of inducing to both brown and white phenotypes20,21. Another limitation of this study was the selective use of growth factors in APC culture. Other growth factor cocktails have been effective in the induction of adipogenesis22. Cell culture in this study was also limited by the fact that all culture was done in two-dimensional culture plates. Although this is the culture norm, cells in vivo do not proliferate in this way. Culturing in three-dimensional environments may allow for further hypertrophy and hyperplasia as it replicates the natural form of the adipose structure23.
Due to the practicality and efficiency of using MCS, this protocol is ideal for the isolation of APC as well as other cell types in PVAT. This procedure also provides a more effective way to induce differentiation in preadipocyte cultures. The minimal cost, equipment, and training required allows this method to be used in any lab wishing to isolate cells based on specific surface markers. Future applications may allow for isolation of other cell populations or use of more specific cell markers. Use of growth factor cocktails in cell commitment may be useful in stem cell activation
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
The Contreras and Watts Laboratories and Dr. William Raphael. These experiments were supported by NHLBI F31 HL128035-01 (tissue digestion protocol standardization), NHLBI 5R01HL117847-02 and 2P01HL070687-11A1 (animals), and NHLBI 5R01HL117847-02 (cell isolation and culture).
Materials
| Tissue Dissection | |||
| Dissecting Dishes | Handmade with Silicone | ||
| Culture Petri Dish | Pyrex | 7740 Glass | |
| Silicone Elastomer | Dow Corning | Sylgard 170 | Kit |
| Braided Silk Suture | Harvard Apparatus | 51-7615 | SP104 |
| Stereomicroscope MZ6 | Leica | 10447254 | |
| Stereomaster Microscope Fiber-Optic Light Source | Fisher Scientific | 12562-36 | |
| Vannas Scissors | George Tiemann & Co | 160-150 | |
| Splinter Forceps | George Tiemann & Co | 160-55 | |
| Tissue Scissors | George Tiemann & Co | 105-400 | |
| KRBB Solution | |||
| Sodium Chloride | Sigma-Aldrich | 7647-14-5 | |
| Potassium Chloride | Sigma-Aldrich | 7447-40-7 | |
| Magnesium Sulfate | Sigma-Aldrich | 7487-88-9 | |
| Potassium Phosphate Dibasic | Sigma-Aldrich | 7758-114 | |
| Glucose | Sigma-Aldrich | 50-99-7 | |
| Antibiotic/Antimicotic | Corning | 30-004-CI | |
| HEPES | Corning | 25-060-CI | |
| Tissue Digestion | |||
| Collagenase Type 1 | Worthington Biochemical | LS004196 | |
| Bovine Serum Albumin | Fisher Scientific | 9048-46-8 | |
| Red Blood Cell Lysis Buffer | BioLegend | 420301 | 1X Working Solution |
| Water Bath | Thermo-Fisher Scientific | 2876 | Reciprocal Shaking Bath |
| Biosafety Cabinet | Thermo-Fisher Scientific | 1385 | |
| Rotisserie Incubator | Daigger | EF4894C | |
| 100 µm Cell Strainer | Thermo-Fisher Scientific | 22-363-549 | Yellow |
| 40 µm Cell Strainer | Thermo-Fisher Scientific | 22-363-547 | Blue |
| Hemocytometer | Cole-Parmer | UX-79001-00 | |
| Trypan Blue | Sigma-Aldrich | 93595 | |
| Cell Isolation | |||
| OctoMACS Kit | Miltenyi Biotech | 130-042-108 | |
| (DMEM):F12 Medium | Corning | 90-090 | Medium Base |
| Fetal Bovine Serum | Corning | 35016CV | USA Origins |
| Normal Donkey Serum | AbCam | AB7475 | |
| Anti-CD34 | Santa Cruz | SC-7324 | FITC conjugated |
| Anti-PDGFRα | Thermo-Fisher Scientific | PA5-17623 | |
| Donkey Anti-Rabbit IgG | Jackson ImmunoResearch | 712-007-003 | |
| PBS 10X | Corning | 46-013-CM | 1X Working Solution |
| EDTA | Fisher Scientific | 15575020 | |
| Cell Culture | |||
| CO2 Cell Incubator | Thermo-Fisher Scientific | 51030285 | Heracell 160i |
| 6-Well Plates | Corning | 3516 | TC-Treated |
| 48- Well Plates | Corning | 3548 | TC-Treated |
| 96-Well Plates, Black Wall | Corning | 353376 | TC-Treated |
| Sodium Bicarbonate | Sigma-Aldrich | 144-55-8 | TC-Treated |
| Fetal Calf Serum | Corning | 35011CV | USA Origins |
| Ascorbic Acid | Sigma-Aldrich | 50-81-7 | |
| Biotin | Sigma-Aldrich | 58-85-5 | |
| Pantothenate | Sigma-Aldrich | 137-08-6 | |
| L-Glutamine | Corning | 61-030 | |
| Bone Morphogenic Protein 4 | Prospec Bio | CYT-081 | |
| Epidermal Growth Factor | PeproTech | 400-25 | |
| Leukemia Inhibitory Factor | PeproTech | 250-02 | |
| Platelet-derived Growth Factor BB | Prospec Bio | CYT-740 | |
| Basic Fibroblast Growth Factor | PeproTech | 450-33 | |
| Insulin | Corning | 25-800-CR | ITS Solution |
| IBMX | Sigma-Aldrich | 28822-58-4 | |
| Dexamethasone | Sigma-Aldrich | 50-02-2 | |
| T3 (Triiodothyronine) | Sigma-Aldrich | 6893-023 | |
| Cell Analysis | |||
| CyQUANT Proliferation Assay | Thermo-Fisher Scientific | C7026 | |
| AdipoRed Fluorescence Assay Reagent | Lonza | PT-7009 | |
| Oil Red O Lipid Dye Reagent | Sigma-Aldrich | O1391 | In Solution |
| M1000 Microplate Reader | Tecan | ||
| Eclipse Inverted Microscope | Nikon | ||
| Digital Sight DS-Qil Camera | Nikon |
Referanslar
- Watts, S. W., et al. Chemerin connects fat to arterial contraction. Arterioscler Thromb Vasc Biol. 33 (6), 1320-1328 (2013).
- Dodson, M. V., et al. Adipose depots differ in cellularity, adipokines produced, gene expression, and cell systems. Adipocyte. 3 (4), 236-241 (2014).
- Police, S. B., Thatcher, S. E., Charnigo, R., Daugherty, A., Cassis, L. A. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 29 (10), 1458-1464 (2009).
- Ruan, H., Zarnowski, M. J., Cushman, S. W., Lodish, H. F. Standard isolation of primary adipose cells from mouse epididymal fat pads induces inflammatory mediators and down-regulates adipocyte genes. J Biol Chem. 278 (48), 47585-47593 (2003).
- Stacey, G. . in eLS. , (2001).
- Swim, H. E., Parker, R. F. Culture characteristics of human fibroblasts propagated serially. Am J Hyg. 66 (2), 235-243 (1957).
- Van Harmelen, V., Rohrig, K., Hauner, H. Comparison of proliferation and differentiation capacity of human adipocyte precursor cells from the omental and subcutaneous adipose tissue depot of obese subjects. Metabolizma. 53 (5), 632-637 (2004).
- Roncari, D. A., Lau, D. C., Kindler, S. Exaggerated replication in culture of adipocyte precursors from massively obese persons. Metabolizma. 30 (5), 425-427 (1981).
- Church, C. D., Berry, R., Rodeheffer, M. S. Isolation and study of adipocyte precursors. Methods Enzymol. 537, 31-46 (2014).
- Fontana, L., Eagon, J. C., Trujillo, M. E., Scherer, P. E., Klein, S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes. 56 (4), 1010-1013 (2007).
- Tchkonia, T., et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 55 (9), 2571-2578 (2006).
- Macotela, Y., et al. Intrinsic differences in adipocyte precursor cells from different white fat depots. Diabetes. 61 (7), 1691-1699 (2012).
- Contreras, G. A., Thelen, K., Ayala-Lopez, N., Watts, S. W. The distribution and adipogenic potential of perivascular adipose tissue adipocyte progenitors is dependent on sexual dimorphism and vessel location. Physiol Rep. 4 (19), (2016).
- Lee, Y. H., Petkova, A. P., Granneman, J. G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 18 (3), 355-367 (2013).
- Lee, Y. H., Petkova, A. P., Mottillo, E. P., Granneman, J. G. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 15 (4), 480-491 (2012).
- Ahrens, M., et al. Expression of human bone morphogenetic proteins-2 or -4 in murine mesenchymal progenitor C3H10T1/2 cells induces differentiation into distinct mesenchymal cell lineages. DNA Cell Biol. 12 (10), 871-880 (1993).
- Bowers, R. R., Lane, M. D. A role for bone morphogenetic protein-4 in adipocyte development. Cell Cycle. 6 (4), 385-389 (2007).
- Schulz, T. J., Tseng, Y. H. Emerging role of bone morphogenetic proteins in adipogenesis and energy metabolism. Cytokine Growth Factor Rev. 20 (5-6), 523-531 (2009).
- Choi, J. R., et al. In situ normoxia enhances survival and proliferation rate of human adipose tissue-derived stromal cells without increasing the risk of tumourigenesis. PLoS One. 10 (1), 0115034 (2015).
- Gupta, R. K., et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 15 (2), 230-239 (2012).
- Rodeheffer, M. S., Birsoy, K., Friedman, J. M. Identification of white adipocyte progenitor cells in vivo. Cell. 135 (2), 240-249 (2008).
- Scott, M. A., Nguyen, V. T., Levi, B., James, A. W. Current methods of adipogenic differentiation of mesenchymal stem cells. Stem Cells Dev. 20 (10), 1793-1804 (2011).
- Edmondson, R., Broglie, J. J., Adcock, A. F., Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. 12 (4), 207-218 (2014).
