This protocol demonstrated the assembly of three chromoprotein cassettes (cjBlue (BBa_K592011), eforRed (BBa_K592012), and amilGFP (BBa_K592010)). First, the coding sequences of the three abovementioned genes and terminators were individually cloned into a C-Brick standard vector. Short DNA parts, promoter and terminator, were introduced into the C-Brick vector through PCR amplification using primers containing the short DNA parts on the 5' terminal. This was followed by the self-ligation method. Three chromoprotein-encoding sequences were first de novo synthesized and then cloned into the C-Brick standard vector through seamless assembly. The primers were described in a previous study4.
After that, cjBlue, eforRed, and amilGFP sequences were ligated with terminator and promoter sequences in the same procedure shown in Figure 2. Correct constructs were transformed into E. coli, demonstrating beautiful colors (Figure 3a). Subsequently, two color-expression cassettes were further assembled with the C-Brick standard to create more colors (i.e., the red color from amilGFP plus eforRed, the green color from amilGFP plus cjBlue, and the light purple color from eforRed plus cjBlue; Figure 3b).
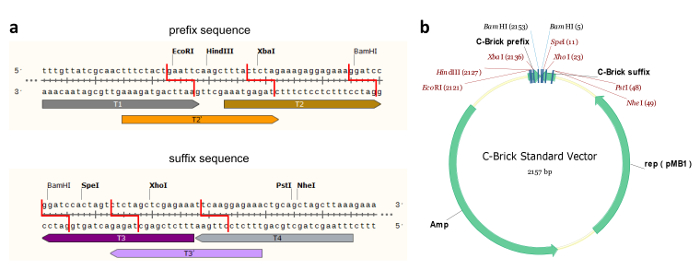
Figure 1: C-Brick Standard Interface DNA Sequence and C-Brick Standard Vector. (a) The sequence of the C-brick standard interface-T1, T2, T3, T4, T2', T3'-can be recognized by its corresponding crRNA and then introduced into a 5' overhang using Cpf1. The cleavage of T2 and T3 (or T2' and T3') sites produces complementary overhangs. With the eight restriction sites designed, the C-Brick standard is partially compatible with BioBrick and BglBrick. (b) Plasmid map for the C-Brick standard vector with restriction digestion sites. Please click here to view a larger version of this figure.
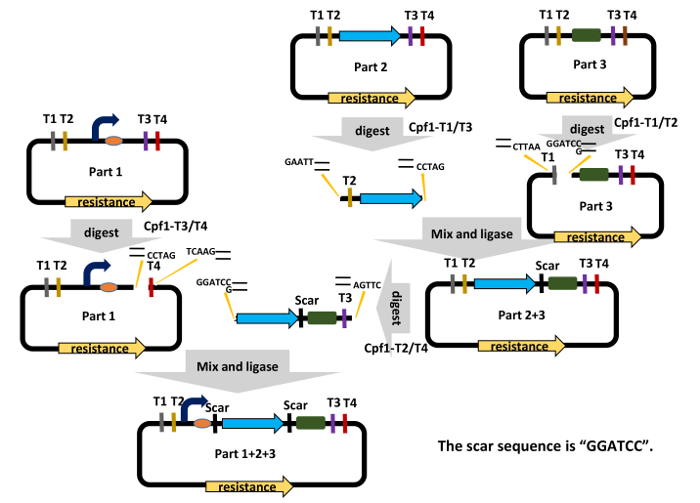
Figure 2: The Workflow for DNA Assembly in the C-Brick Standard. Cpf1-digested T2 and T3 target sites produce complementary cohesive ends of "GGATC" and "GATCC," respectively, which can be ligated to generate a short "GGATCC" scar. Detailed procedures can be found in step 3 in the text ("C-Brick assembly"). Please click here to view a larger version of this figure.
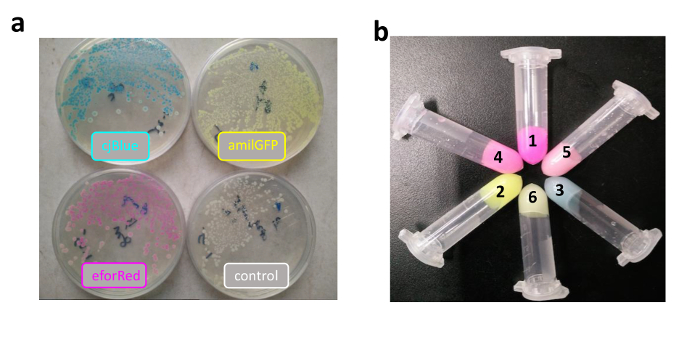
Figure 3: Colorful Bacterial Pigments Produced by E. coli Harboring Constructs Assembled in the C-Brick Standard. (a) Three chromoproteins were expressed in E. coli grown on a plate. Bacteria with no chromoproteins (negative control) are shown on the 4th plate. (b) Three chromoproteins and three assembled dual chromoproteins were expressed in E. coli grown in liquid LB medium. From 1 to 6, the constructs expressed eforRed (1), amilGFP (2), cjBlue (3), amilGFP plus eforRed (4), amilGFP plus cjBlue (5), and eforRed plus cjBlue (6). The colorful bacteria on a single plate or in a single microcentrifuge tube were cultured from a single sequence-verified clone. Please click here to view a larger version of this figure.
| İsim | sequence | ||
| T7-F | GAAATTAATACGACTCACTATAGGG | ||
| T7-T1-R | gaattcagtagaaagttgcgataaATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
| T7-T2'-R | ctagagtaaagcttgaattcagtaATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
| T7-T2-R | ggatcctttctcctctttctagagATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
| T7-T3-R | ggatccactagtctctagctcgaATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
| T7-T3'-R | ctctagctcgagaaattcaaggaATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
| T7-T4-R | ttcaagggaaactgcagctagctATCTACAACAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC | ||
Table 1: Oligos for the Preparation of the crRNA Transcription Templates used in this Study. T7-F was the top-strand oligonucleotide, and the others were the bottom-strand oligonucleotides. The lowercase bases in the sequence will be transcribed into the guide sequence in crRNA.
| names | RNA sequences (5'-3') | ||
| crRNA-T1 | GGGAAUUUCUACUGUUGUAGAUUUAUCGCAACUUUCUACUGAAUUC | ||
| crRNA-T2 | GGGAAUUUCUACUGUUGUAGAUCUCUAGAAAGAGGAGAAAGGAUCC | ||
| crRNA-T3 | GGGAAUUUCUACUGUUGUAGAUUCGAGCUAGAGACUAGUGGAUCC | ||
| crRNA-T4 | GGGAAUUUCUACUGUUGUAGAUAGCUAGCUGCAGUUUCUCCUUGAA | ||
| crRNA-T2' | GGGAAUUUCUACUGUUGUAGAUUACUGAAUUCAAGCUUUACUCUAG | ||
| crRNA-T3' | GGGAAUUUCUACUGUUGUAGAUUCCUUGAAUUUCUCGAGCUAGAG | ||
Table 2: The crRNA Sequences used in this Study. The crRNAs were produced through the in vitro transcription in step 1 in the text ("Preparation of crRNA").
