Trehalose is a symmetrical non-reducing disaccharide consisting of two glucose moieties that are joined by a 1,1-α,α-glycosidic bond (Figure 1A). While trehalose is absent from humans and other mammals, it is found commonly in bacteria, fungi, plants, and invertebrates 1. The primary role of trehalose in most organisms is to protect against environmental stresses, such as desiccation 1. In addition, some human pathogens require trehalose for virulence, including the tuberculosis-causing Mycobacterium tuberculosis, which utilizes trehalose as a mediator of cell envelope biosynthesis and as a building block for the construction of immunomodulatory glycolipids 2.
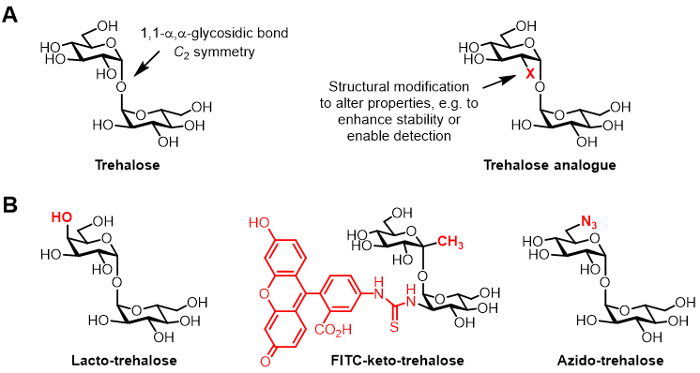
Figure 1: Trehalose and trehalose analogues. (A) Structures of natural trehalose and an unnatural trehalose analogue, where X is a structural modification. (B) Examples of trehalose analogues reported in the literature that have potential applications in biopreservation and bioimaging.
Due to its unique structure and physiological functions, trehalose has drawn significant attention for use in bio(techno)logical and biomedical applications 3. The protective properties of trehalose observed in nature—e.g., its striking ability to help sustain life in "resurrection" plants that have undergone extreme dehydration 4—have spurred its extensive use in biopreservation applications. Trehalose has been used to preserve a wide array of biological samples, such as nucleic acids, proteins, cells, and tissues 3. For instance, trehalose is used as a stabilizing additive in a number of pharmaceuticals that are on the market, including several anti-cancer monoclonal antibodies 3. As well, trehalose is used as a sweetener in the food industry, and it is extensively used for product preservation in both the food and cosmetics industries. The adoption of trehalose for these types of commercial applications was initially limited by the inability to obtain bulk quantities of pure trehalose from natural sources or through synthesis. However, an efficient enzymatic process for the economical production of trehalose from starch has recently been developed, which has spurred its widespread commercial use 5.
Chemically modified derivatives of trehalose, referred to herein as trehalose analogues, have gained increasing attention for various applications (generic structure shown in Figure 1A; specific examples of trehalose analogues shown in Figure 1B) 6. For example, lacto-trehalose is a trehalose analogue with one of its glucose units replaced with galactose, thus its 4-position hydroxyl group has an inverted stereochemical configuration. Lacto-trehalose has the same stabilizing properties as trehalose but is resistant to degradation by intestinal enzymes, making it attractive as a non-caloric food additive 6, 7.
Our group's interest in trehalose analogues primarily relates to their value as mycobacteria-specific probes and inhibitors. The Barry and Davis groups developed a fluorescein-conjugated keto-trehalose analogue, named FITC-keto-trehalose, which was shown to metabolically label the cell wall of live M. tuberculosis, enabling its detection by fluorescence microscopy 8. The Bertozzi lab developed smaller azido-trehalose (TreAz) analogues that could metabolically label the cell wall and subsequently be detected using click chemistry and fluorescence analysis 9. These advances point to the possibility of using trehalose-based probes as diagnostic imaging agents for tuberculosis. Trehalose analogues have also been pursued as inhibitors of M. tuberculosis due to their potential to disrupt pathways in the bacterium that are essential for viability and virulence 10,11,12.
So far, the main obstacle to developing trehalose analogues for bio(techno)logical and biomedical applications is the lack of efficient synthetic methods. The two traditional routes to producing trehalose analogues rely on chemical synthesis (Figure 2). One route involves desymmetrization/modification of natural trehalose, while the other involves starting with properly functionalized monosaccharide building blocks and performing chemical glycosylation to forge the 1,1-α,α-glycosidic bond. These approaches, which have recently been discussed in review articles 13, 14, have proven useful for accomplishing multistep synthesis of small quantities of complex trehalose-containing natural products, such as sulfolipid-1 from M. tuberculosis 15. However, both approaches are generally inefficient, time-consuming, inaccessible to non-chemists, and, additionally, are not considered to be environmentally friendly. Thus, for synthesizing certain types of trehalose analogues, these strategies are not ideal.
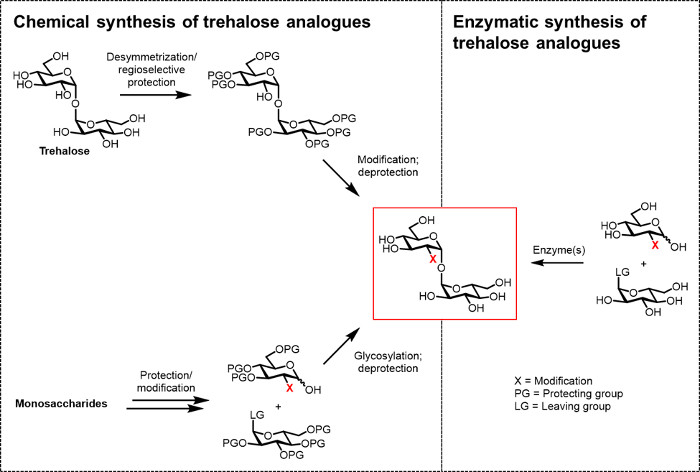
Figure 2: Approaches to trehalose analogue synthesis. Chemical approaches to trehalose analogue synthesis, shown on the left, use multistep procedures that involve difficult protection/deprotection, desymmetrization, and/or glycosylation steps. Enzymatic synthesis, shown on the right, uses enzyme(s) to stereoselectively convert simple, unprotected substrates to trehalose analogues in aqueous solution. The enzymatic protocol reported herein uses a trehalose synthase (TreT) enzyme to convert glucose analogues and UDP-glucose into trehalose analogues in a single step. Please click here to view a larger version of this figure.
An efficient biocatalytic route to trehalose analogues would facilitate the production, evaluation, and application of this promising class of molecules. While the commercial enzymatic process for trehalose production5 is not adaptable to synthesizing analogues because it utilizes starch as a substrate, there are other biosynthetic pathways in nature that may be exploited for trehalose analogue synthesis. However, research in this area, which was recently reviewed 6, has been limited. One report used a method inspired by the Escherichia coli trehalose biosynthetic pathway to access a single fluoro-trehalose analogue from the corresponding fluoro-glucose. However, this approach requires a three-enzyme system that has limited efficiency and generality 8. Another approach that has been explored is to use trehalose phosphorylase (TreP) in the reverse direction, which in principle permits the one-step synthesis of trehalose analogues from glucose analogues and glucose-1-phosphate 6, 16, 17. Although this approach may have future promise, both inverting and retaining TrePs currently have drawbacks for analogue synthesis. For example, inverting TrePs have a prohibitively expensive donor molecule (β-D-glucose 1-phosphate) and retaining TrePs have poor enzyme expression yields/stability and limited substrate promiscuity. Significant improvements (e.g., via enzyme engineering) will be needed before TreP-mediated analogue synthesis is practical.
At present, the most practical approach for the enzymatic synthesis of trehalose analogues is to use a trehalose synthase (TreT) enzyme, which converts glucose and uridine diphosphate (UDP)-glucose into trehalose in a single step 6. We recently reported the use of Thermoproteus tenax TreT—a thermostable and unidirectional enzyme 18—to synthesize trehalose analogues from glucose analogues and UDP-glucose (Figure 3) 19. This enzyme only operates in the synthetic direction and avoids the problem of trehalose degradation found in the TreP system. This one-step reaction could be completed in 1 hour, and a broad variety of trehalose analogues were accessed in high yield (up to >99% as determined by high performance liquid chromatography (HPLC)) from readily available glucose analogue substrates (see Table 1 in the Representative Results section).
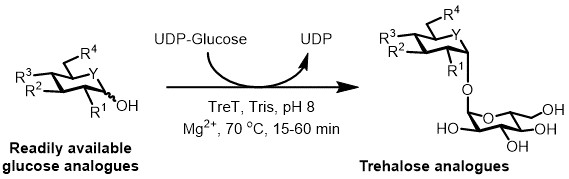
Figure 3: TreT-catalyzed one-step synthesis of trehalose analogues. The TreT enzyme from T. tenax can stereoselectively join readily available glucose analogues and UDP-glucose to form trehalose analogues in one step. R1-R4 = Variable structural modification, for example azido-, fluoro-, deoxy-, thio-, stereochemical, or isotopic label modifications; Y = variable heteroatom, for example oxygen or sulfur, or isotopically labeled heteroatom.
Here, we provide a detailed protocol for the TreT synthesis process, including expression and purification of TreT from E. coli, optimized TreT reaction conditions, and an improved purification method that is carried out entirely in the aqueous phase. This modified protocol enables the expedient and efficient synthesis and purification of diverse trehalose analogues on a semi-preparative scale (10-100 mg). We also demonstrate the use of this protocol for preparing and administering a trehalose-based probe to mycobacteria in less than 1 hour, which enabled the rapid fluorescence detection of mycobacterial cells.
T. tenax TreT was obtained from E. coli in a yield of approximately 4 mg/L using standard protein expression and purification techniques. A single nickel affinity chromatography step was sufficient to purify TreT from E. coli lysate (a representative FPLC trace is shown in Figure 4). As established in our initial publication on the TreT synthesis process, recombinant T. tenax TreT is capable of converting a broad of variety glucose analogues-many of which are commercially available-to the corresponding trehalose analogues with high efficiency 19. Table 1, which gives HPLC-determined reaction yields for a number of starting glucose analogues using our initially reported protocol, illustrates the scope of TreT's substrate promiscuity and its suitability for synthesis. Diverse structural modifications, including fluoro-, deoxy-, azido-, thio-, and stereochemical modifications at different positions around the sugar ring are well-tolerated by the wild-type enzyme, with the main exception being modifications at the 4-position.
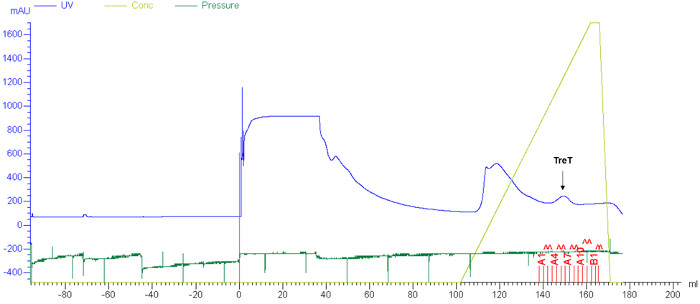
Figure 4: Representative results for TreT purification by FPLC. Purification of recombinant T. tenax TreT from E. coli lysate was carried out using nickel affinity chromatography as described in steps 1.4.1-1.4.4. Blue, ultraviolet (UV) absorbance trace; light green, concentration of elution buffer; dark green, pressure. The peak corresponding to TreT is indicated with an arrow. Please click here to view a larger version of this figure.
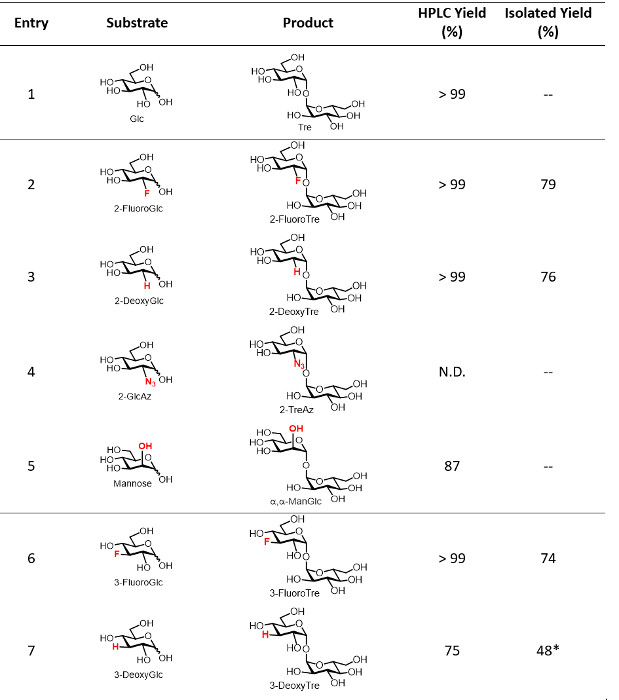
Table 1: Representative yields for the TreT reaction. HPLC-determined and isolated yields of the TreT reaction for several trehalose analogues. N.D., not detected. — Indicates the reaction was not performed on a semi-preparative scale using the optimized protocol. *Indicates that a size exclusion chromatography step was required. Horizontal dividing lines separate the position of modification on the trehalose sugar ring. Table adapted from reference 19 with permission; updated to include isolated yields from this work. Please click here to download this file.
Herein, we report optimizations to our initial protocol that improve the overall efficiency and speed of the TreT synthesis process. While the original protocol used 10 mM glucose analogue and 40 mM UDP-glucose, it was determined that comparable conversion could be obtained using 20 mM glucose and 40 mM UDP-glucose, effectively doubling the amount of product generated per reaction and limiting the waste of UDP-glucose, which is relatively expensive. Using equimolar amounts of glucose analogue and UDP-glucose resulted in lower conversions. If desired, reaction times can also be shortened to less than the initially reported 60 min while still retaining comparable conversion for many analogues, which was demonstrated by the quantitative conversion of 6-GlcAz to 6-TreAz in only 15 min at 70 °C (Figures 5 and 6).
The purification protocol reported herein is substantially improved, replacing the original enzyme precipitation/silica gel chromatography method with a non-chromatographic spin dialysis/ion exchange method. The rationale for this modification is that the TreT reaction mixture consists entirely of ionic species except for the neutral trehalose analogue product. Therefore, the mixture can be spin-dialyzed to remove enzyme and then treated with mixed-bed ion exchange resin to remove all of the ionic species, delivering purified trehalose analogue in aqueous solution in as little as 45 min. This all-aqueous purification method avoids environmentally detrimental organic solvents, time-consuming evaporation and filtration steps, and dry-load silica gel chromatography, which is a slow and cumbersome process, especially for non-chemists. For TreT reactions that exhibit quantitative conversion as determined using the described TLC or HPLC analyses, this purification method provides isolated yields of approximately 60-80% at the reported reaction scale, with the product loss likely due to binding of some sugar to the ion exchange resin (see Table 1 for representative isolated yields). In cases where unreacted glucose analogue remains, it can be separated from the desired trehalose analogue product using a polyacrylamide bead-based size exclusion column, eluting with water. If preferred, this could also be accomplished using preparative-scale HPLC with an aminopropyl column. Product purity can be assessed by TLC, HPLC, and/or NMR spectroscopic analysis. Figure 5 shows representative analytical data for 6-TreAz, which was synthesized via the described protocol. The TLC and HPLC data indicated quantitative conversion of 6-GlcAz substrate to 6-TreAz product for this reaction, and the isolated yield after the spin dialysis/ion exchange purification steps was 58%. 1H and 13C NMR spectroscopic analysis confirmed the structure of the 6-TreAz product, most notably including α,α-stereochemistry of the newly formed glycosidic bond, as well as its purity.
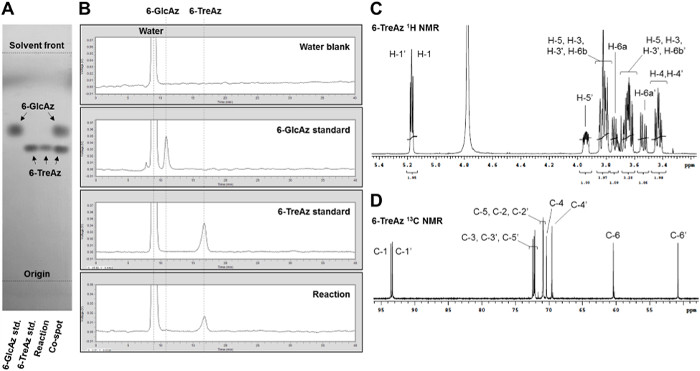
Figure 5: Representative analytical data for the TreT reaction. (A) TLC and (B) HPLC analysis of TreT-catalyzed conversion of 6-GlcAz to 6-TreAz. (C) 1H NMR and (D) 13C NMR spectra of the 6-TreAz product obtained after spin dialysis/ion exchange purification. Please click here to view a larger version of this figure.
Given the aforementioned value of trehalose analogues for the specific detection of M. tuberculosis, the TreT process should facilitate the development and use of trehalose-based probes for tuberculosis research and diagnostic applications. To demonstrate this type of application, we used the optimized TreT process to rapidly prepare, purify, and administer 6-TreAz—a trehalose-based click chemistry probe 9—to mycobacteria to enable fluorescence detection (workflow and representative data are shown in Figure 6). Briefly, TreT was used to convert commercial 6-GlcAz to 6-TreAz quantitatively in 15 min, and then the reaction mixture was subjected to the spin dialysis/ion exchange purification method, which took only 45 min (multiple rinse steps were omitted to increase speed). Thus, an aqueous stock solution of pure 6-TreAz of known concentration (~5 mM) was generated in only 1 hour. The 6-TreAz stock was immediately administered to a growing culture of the model bacterium Msmeg (final 6-TreAz concentration ~25 µM) to accomplish azide labeling of the cell surface, which embedded a handle for click chemistry-mediated ligation 20,21 of a fluorescent probe. Subsequently, labeled cells were analyzed by fluorescence microscopy, which showed strong fluorescence for 6-TreAz-treated cells. As expected, control samples that were not treated with 6-TreAz or that were missing a functional trehalose transporter 22, which is required for 6-TreAz uptake and metabolic incorporation 9, showed no fluorescence.
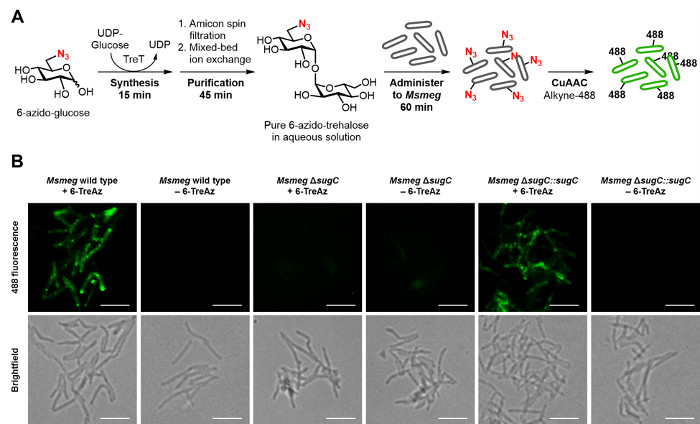
Figure 6: Representative results for the rapid detection of mycobacteria using a TreT-synthesized trehalose analogue. (A) Workflow for the rapid synthesis, purification, and use of 6-TreAz for fluorescence detection of Msmeg. Steps 5.1.1-5.1.6 of the protocol were used to synthesize and purify 6-TreAz (giving a ~5 mM aqueous solution in 1 hour), then administer it to live Msmeg to accomplish metabolic labeling of the cell surface with azides. Next, as described in steps 5.2.1-5.2.9, click chemistry (CuAAC) was performed to react cell-surface azides with an alkyne-modified fluorophore, alkyne-488. (B) Fluorescence imaging of 6-TreAz-treated Msmeg containing a functional trehalose transporter (wild type and ΔsugC::sugC) showed strong fluorescence, while the control samples of Msmeg left untreated or lacking the trehalose transporter (ΔsugC) did not. Figure adapted from reference 19 with permission; updated with workflow and imaging data from the improved TreT protocol. Scale bars, 5 µm. Please click here to view a larger version of this figure.
