The described procedure allows isolation of approximately 12 x 107 Sertoli cells from 10 rat testes. 3 x 106 cells are plated per well on a 6-well plate so that six to seven 6-well plates are available for experiments on day 7. The Trypsin-DNase I digestion is the most critical step during Sertoli cell isolation. If digestion at this point is advancing too far, the ratio of germ cells/Sertoli cells will disproportionally increase up to the end. During day 3-6 of culture it is important to carefully wash away as many non-adhering or loosely attached germ cells as possible. As an alternative to extensive washings over 4 days germ cells can be removed by hypotonic treatment on day 327. An advantage of this method is that the time of Sertoli cell culture is kept to a minimum so that Sertoli cell-specific functions are potentially better retained than after a prolonged period of culture.
Although the Hyaluronidase treatment serves to remove most residual peritubular cells, a few cells will survive the treatment. Purity of the isolated Sertoli cells is >95% as can be demonstrated by vimentin immunolabeling (Figure 5B). Because peritubular cells have a strong proliferative potential, Sertoli cell culture has to be performed in the absence of serum. In the presence of 10% serum PTCs would constitute approximately 20% of all cells after 6 days of culture whereas its fraction can be expected to be below 1% in the absence of serum7. Virtually all isolated Sertoli cells are viable as judged by Trypan blue staining (not shown). Around day 7 of culture the isolated Sertoli cells are least contaminated with germ cells and peritubular cells (Figure 4), so that experiments should be performed around this day if possible. For good reproducibility it is important to repeat experiments always on the same day of culture. If transfections are planned they should be performed on day 4 or 5, and cell extracts should be made on day 5 or 6.
Over at least two weeks in serum-free conditions Sertoli cells are responsive to FSH and produce more androgen-binding protein, plasminogen activator and transferrin upon FSH treatment. For longer culture a feeder layer of peritubular cells or serum is required. After four weeks cultures deteriorate, and cells detach from the substratum7.
On day 6 of culture most Sertoli cells have acquired lipid droplets. In the majority of cells a single large vacuole has formed (Figure 5C and 5D). The neutral lipid content can be easily visualized by Oil Red O staining (Figure 4F).
In addition to Sertoli cells the removed PTCs can be cultured as well (Figure 4A and 4B). PTCs from 10 testes yield five T75 flasks that can be expected to form a confluent layer after 2 days of culture. Like isolated Sertoli cells freshly isolated PTCs are contaminated by germ cells that can be largely removed by passaging. Flasks are split 1:2 by standard trypsinization so that 10 flasks will be confluent on day 5 after isolation. Purity of isolated PTCs is >95% and can be confirmed using α-smooth muscle actin immunolabeling (Figure 5A). From this day onwards PTCs can be used for experiments. One flask is sufficient for approximately one 6-well plate, and wells should be confluent the next day. Cells can be passaged many times, but experiments should be performed always at the same passage in order to make sure that cells behave in a reproducible manner.
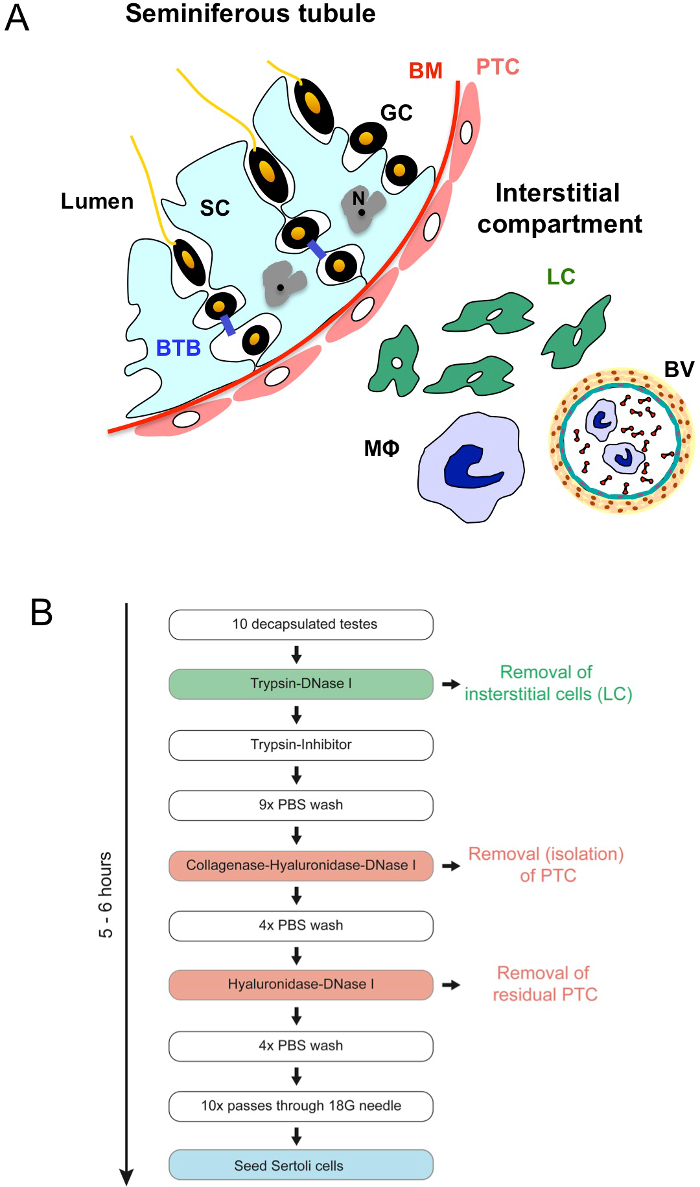
Figure 1. Morphology of the testis and workflow of the procedure. (A) A sector of a seminiferous tubule with adjacent interstitial tissue is schematically shown. Seminiferous tubules are enclosed by a basal membrane (BM) and a circumferential layer of myoid peritubular cells (PTC). The germinal epithelium is located on the luminal side of the basal membrane. Sertoli cells (SC), spanning the whole germinal epithelium are strongly attached to the BM and interconnected via basolateral positioned occluding junctions representing the blood-testis barrier (BTB). Their irregular nucleus (N) shows one or more fissure-like indentations and contains a nucleolus. Germ cells (GC) are in intimate contact with SCs at all stages of their development. The interstitial compartment between the tubules contains Leydig cells (LC) and immune cells such as macrophages (MΦ) as well as blood vessels (BV). Figure adapted from Fijak and Meinhardt28. (B) Workflow of the procedure; color coding is like in (A). Please click here to view a larger version of this figure.
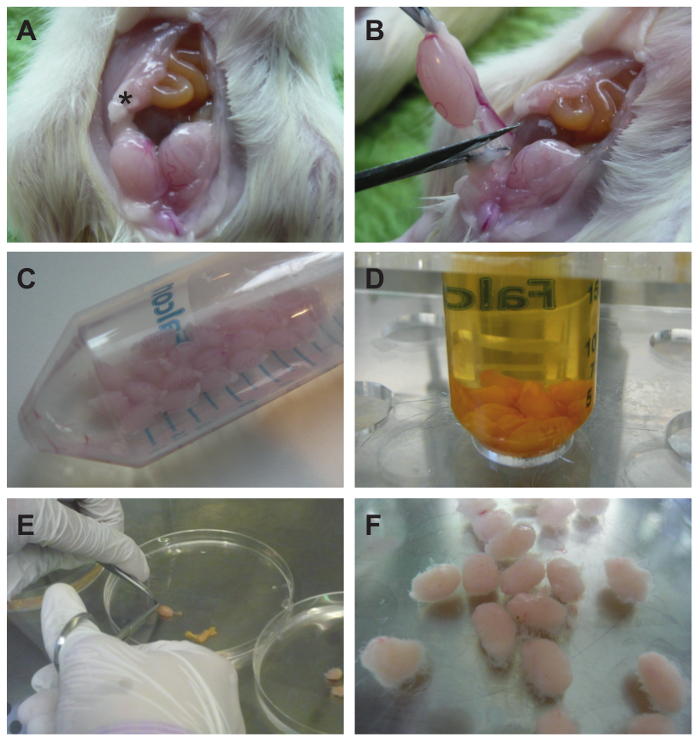
Figure 2. Testes from Wistar rats are excised and decapsulated. (A) The peritoneal cavity is opened through a longitudinal incision as described in the text. An asterisk (*) indicates the epidydimal fat pad. (B) Testes are removed by cutting the spermatic cord and (C) collected in a 50 ml conical tube containing 20 ml PBS. When all testes have been collected they are disinfected in 20 ml of 1% (w/v) iodine in ethanol. For removal of the iodine they are quickly washed twice with 25 ml PBS each. (D) Testes after first PBS wash. (E) Testes are transferred to a Petri dish (on the right), and seminiferous tubules are squeezed out of the opened tunica albuginea by means of closed scissor blades (on the left) as described in the text. (F) The decapsulated testes still form a compact testicle-shaped mass with single tubules protruding. Please click here to view a larger version of this figure.
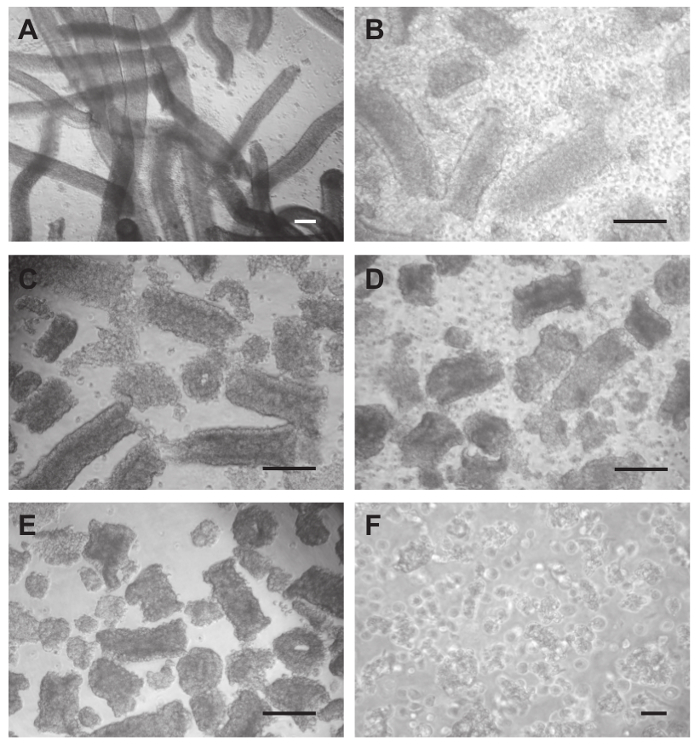
Figure 3. Isolation of Sertoli cells. (A) After removal of interstitial cells by Trypsin-DNase I digestion and washing 9x with PBS tubules become mobilized and look clean. (B) During collagenase-hyaluronidase-DNase I treatment peritubular cells from the outer layer and germ cells are released. The tubules get shortened and obtain a rough appearance. (C) Tubular fragments after washing 4x with PBS. (D) Hyaluronidase-DNase I treatment releases residual peritubular cells as well as germ cells. Tubules get further shortened, and tubular aggregates form. (E) Tubular aggregates after washing 4x with PBS. (F) Single Sertoli cells are produced by passing the aggregates 10x through an 18G needle. Scale bars in (A-E) = 200 µm, in (F) = 50 µm. Please click here to view a larger version of this figure.
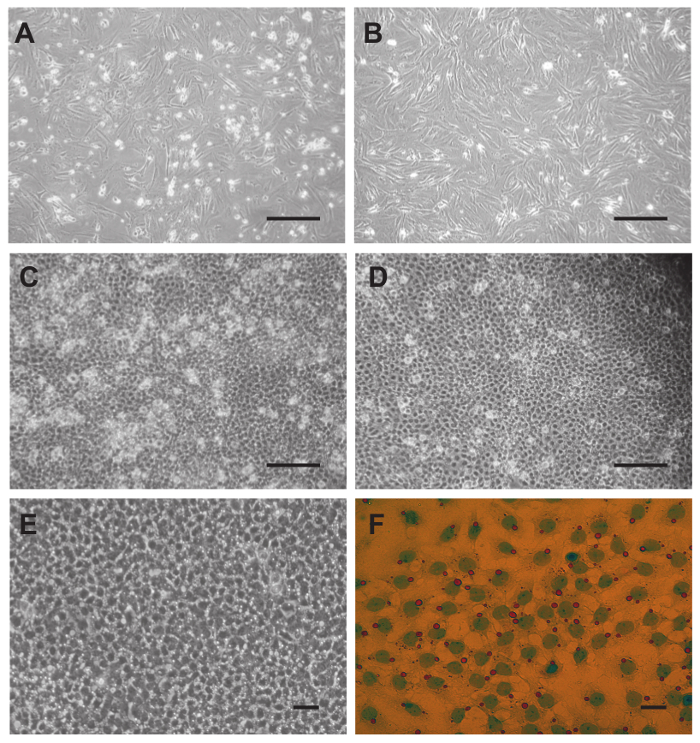
Figure 4. Culture of peritubular cells (A-B) and Sertoli cells (C-F) and removal of contaminating germ cells. (A) Resuspended PTCs from step 4.5 were seeded immediately and photographed the next day. (B) After splitting on day 3 contamination with germ cells is reduced. (C) On day 4 Sertoli cells show a typical cobblestone-like pattern. Floating and adhering germ cells are visible before washing with PBS. (D) The same well after washing 3x with PBS. The number of contaminating germ cells is reduced. Washing thrice with PBS is repeated on day 5 and 6. (E) On day 6 of culture virtually all germ cells have been removed. Sertoli cells have formed a unique looking cell layer, and most cells have acquired a single large vacuole. (F) Oil red O staining shows that these vacuoles are lipid droplets containing largely neutral lipids. Scale bars in (A-D) = 200 µm, in (E) = 50 µm and in (F) = 25 µm. Please click here to view a larger version of this figure.
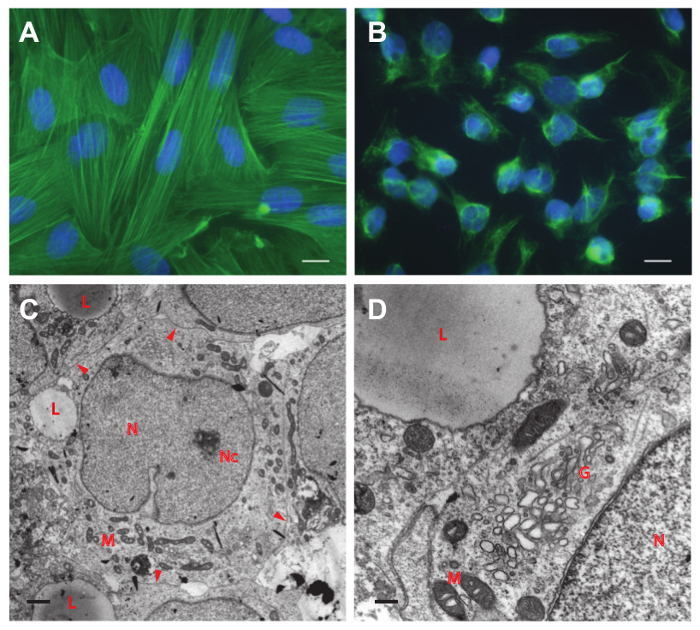
Figure 5. Immunofluorescence staining. Staining of purified primary peritubular cells with actin (smooth muscle) antibody (A) and of Sertoli cells with vimentin antibody (B). No contaminating cells are visible in both fields of view. Scale bar in (A) and (B) = 10 µm. (C) and (D) Electron microscopic pictures of Sertoli cells after 4 days in culture. (C) Note the close contact of adjacent cells (arrowheads) that leads to the cobblestone-like pattern as seen in Figure 4C. A single lipid droplet (L) is observed in the cytoplasm of each cell. (D) Most abundant organelles are mitochondria (M) and the Golgi apparatus (G). The nucleus (N) contains a nucleolus (Nc) and shows the typical fissure-like indentation. Scale bar in (C) = 1 µm and in (D) = 0.25 µm. Please click here to view a larger version of this figure.
