Quality control of the decellularization procedure
The efficiency of the decellularization can be monitored by analyzing the protein content of each fraction by western blot. Table 2 lists proteins of diagnostic value to assess the quality of the decellularization procedure. Figure 2A shows the efficient extraction in the intermediate fractions of cytosolic (GAPDH), nuclear (histones), membrane (β1 integrin) and cytoskeletal (actin) proteins, whereas no ECM proteins (collagen I) is detected in these fractions (Figure 2). In turn, the final pellet is enriched for ECM proteins and largely depleted of intracellular proteins (Figure 2A). Figure 2B presents satisfactory intracellular protein depletion (no histone is detected in the ECM-rich fraction), although, actin can still be detected in the ECM-rich fraction and depletion of monomeric collagen I — apparent molecular weight ~110 kDa, presumptively corresponding to unassembled collagen I — can be observed in the CS fraction.
We also routinely monitor additional ECM proteins such as fibronectin and laminin, although, in some tissues these components can be partially solubilized in earlier fractions8-10. For example, fibronectin also occurs as soluble plasma fibronectin that is not incorporated into the ECM. Perfusion of the tissue prior to extraction reduces plasma fibronectin concentration but does not always eliminate it. In some tissues, laminins are found loosely associated with the cell surface or the ECM and extract in intermediate fractions. If this occurs it can be addressed by altering extraction conditions (see Discussion).
Note that the enrichment of ECM proteins and concomitant depletion of intracellular components is based on the relative solubility of proteins in the different buffers. This differs among different tissues — in some cases the histones and actin are more readily extracted than in others. Also note that, although histones are expected to be extracted in the N fraction (Figure 2B), we often observe a more complete depletion of histones in the M or CS fraction (Figure 2A and B).
Indicative peptide concentration expected
The concentration of the peptide solution obtained after digestion, acidification and desalting can be measured by spectrophotometry either by measuring the absorbance of the peptide solution using the 280 nm wavelength corresponding to tryptophan, tyrosine, or using the 205 nm wavelength corresponding to absorbance of the peptide bonds.
We measured the concentration of the peptide solutions obtained from the decellularization of three murine lungs samples (82 mg, 100 mg, and 100 mg, respectively) prepared in parallel and obtained from each: 424 ng/µl, 450 ng/µl, and 580 ng/µl of peptides respectively.
Identification of ECM peptides by mass spectrometry
Mass spectrometric analysis of the composition of ECM-enriched protein samples, prepared as described here, showed that >70% of the signal intensity corresponds to ECM and ECM-associated proteins8-10.
Table 1. Volume of reagents from Compartmental Extraction kit to decellularize 100 mg (wet weight) of tissue or tumor. This table lists the composition and the volume of each buffer used to conduct the decellularization of 100 mg of tissue or tumor. A cocktail of protease inhibitors is provided as a 50x solution and needs to be added to each buffer2.
| Reagents from Compartment Protein Extraction Kit | Volume for 100 mg of tissue | Composition (based on Millipore datasheet cat#21451) |
| Buffer C | 500 μl | HEPES (pH 7.93), MgCl2, KCl, EDT4, Sucrose, Glycerol, Sodium Orthovanadate5 |
| Buffer W | 400 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| Buffer N | 150 μl x 2 | HEPES (pH 7.9), MgCl2, NaCl, EDTA, Glycerol, Sodium Orthovanadate |
| Buffer W | 400 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| Buffer M | 100 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium deoxycholate (DOC)6, NP-406, Sodium Orthovanadate |
| Buffer CS | 200 μl | PIPES (pH 6.8), MgCl2, NaCl, EDTA, Sucrose, Sodium Dodecyl Sulfate (SDS)7, Sodium Orthovanadate |
| Buffer C | 150 μl | HEPES (pH 7.9), MgCl2, KCl, EDTA, Sucrose, Glycerol, Sodium Orthovanadate |
| 1x PBS | 500 μl/wash | – |
Notes:
1 For proprietary reasons, we were unable to obtain the precise composition of the buffers from the supplier of the kit, but we include here some notes based on our own experience using home-made buffers to conduct similar extractions.
2 Protease inhibitor: it is advisable to include a variety of inhibitors against cysteine, serine and threonine peptidases, serine esterases, divalent cation-dependent metalloproteinases etc. Many commercially available protease inhibitor cocktails exist.
3 pH above 7.0 is an effective inhibitor of lysosomal proteases.
4 EDTA (usually used at 2 mM) is an effective inhibitor of divalent cation-dependent proteases.
5 Sodium orthovanadate is a phosphatase inhibitor. A typical effective concentration would be 0.5–5 mM.
6 NP40 at 0.1%–0.5% is sufficient to solubilize most membrane lipids. The combination of NP40 and DOC — often used at equal concentrations (e.g., 0.5% of each) — is often used as a more stringent extraction that still leaves many protein-protein interactions intact.
7 SDS is a more stringent ionic detergent (CMC 0.1%). It can also be used in combination with the other two detergents SDS/NP40/DOC 0.1/0.5/0.5% as an intermediate stringency buffer.
Table 2. Diagnostic proteins to monitor the quality of the decellularization procedure. This table lists examples of proteins that are characteristics of each subcellular compartment (cytosol, nucleus, plasma membrane, cytoskeleton and ECM) that can be used to monitor the quality of the decellularization procedure and the efficiency of the ECM-enrichment.
| Intracellular Compartment | Diagnostic Proteins |
| Cytosolic proteins | Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) |
| Nuclear proteins | Histones, Lamins, Nucleoporin |
| Membrane proteins | Integrins, Transferrin Receptor |
| Cytoskeletal proteins | Actin, Tubulin, Vimentin |
| Basement membrane ECM proteins | Collagen IV, Nidogens, Laminins |
| Interstitial ECM proteins (interstitial) | Collagen I, Collagen III, Collagen VI, Fibronectin |
For recommendation on antibodies, see our publications8-10.
Table 3. Volume of reagents to digest ECM-enriched samples into peptides. This table lists the reagents used to resuspend ECM-enriched protein samples and reduce, alkylate, deglycosylate and digest proteins samples into peptides prior to mass spectrometry analysis.
| Reagents | Preparation | Final Concentration / Amount for 1 mm-thick pellet (~5–10 mg dry weight) | Volume for 1 mm-thick pellet (~5–10 mg dry weight) |
| Ammonium bicarbonate (NH4HCO3) | 100 mM solution in HPLC-grade water | - | - |
| Urea | 8 M solution in 100 mM ammonium bicarbonate | 8 M | 50 μl |
| Dithiothreitol | Reconstitute in HPLC-grade water at 500 mM | 10 mM | 1 μl |
| Iodoacetamide | Reconstitute in HPLC-grade water at 500 mM | 25 mM | 2.5 μl |
| Peptide-N-Glycosidase F (PNGaseF) | Commercial solution at 500 U/μl | 1,000 U | 2 μl |
| Endoproteinase LysC, mass spectrometry-grade | Reconstitute in HPLC-grade water at 0.5 μg/μl | 1 μg | 2 μl |
| Trypsin, mass spectrometry-grade (round 1) | Commercial solution at 0.5 μg/μl | 3 μg | 6 μl |
| Trypsin, mass spectrometry-grade (round 2) | Commercial solution at 0.5 μg/μl | 1.5 μg | 3 μl |
| Trifluoro-acetic acid (TFA) | 50% solution in HPLC-grade water | - | 2–5 μl |
| Acetonitrile (elution) | 60% solution with 0.1% TFA in HPLC-grade water | - | 500 μl |
| Acetonitrile (reconstitution) | 3% solution with 0.1% TFA in HPLC-grade water | - | 100 μl |
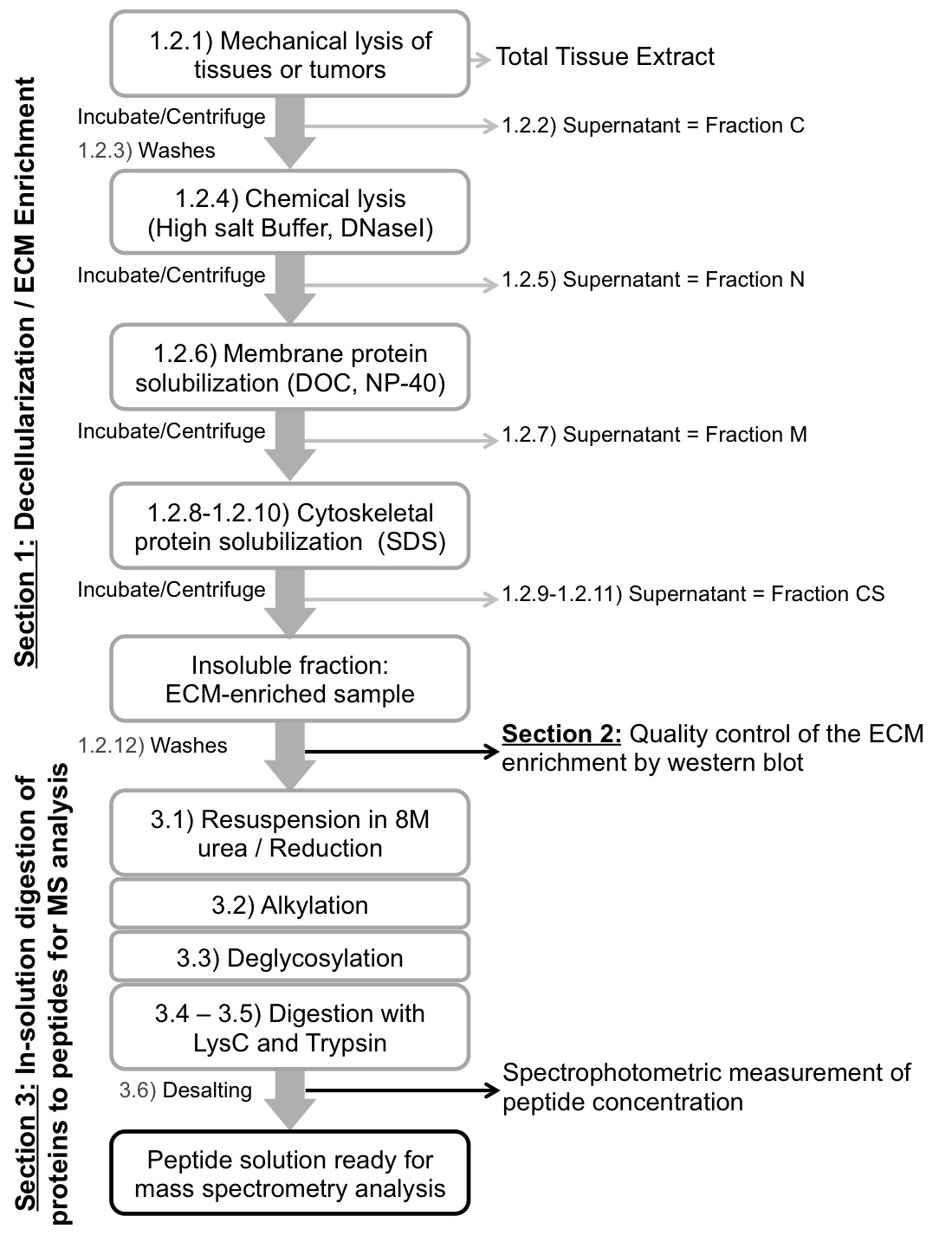
Figure 1. Scheme of the experimental procedure. Schematic workflow of the protocol to decellularize tissues (Section 1), control the quality of the decellularization and evaluate the ECM enrichment (Section 2) and digest ECM-enriched protein samples into peptides prior to mass spectrometry analysis (Section 3).
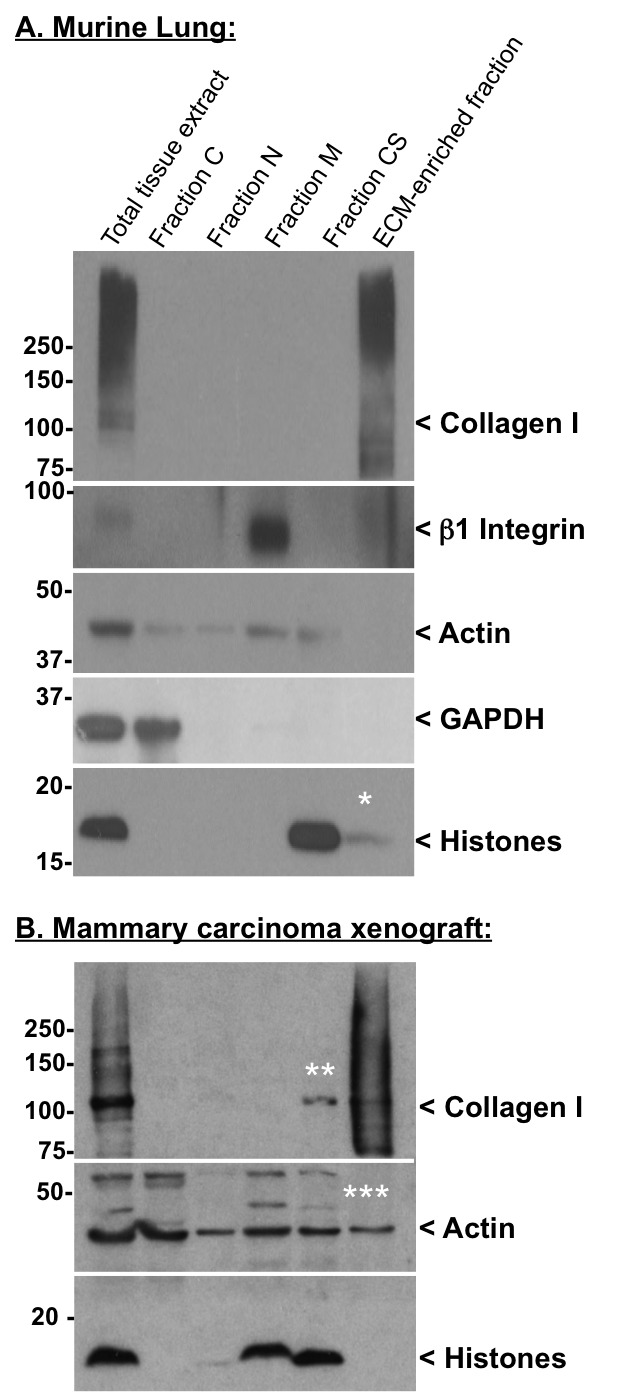
Figure 2. Quality control of the decellularization procedure by western blot. Western blots were performed on murine lung (A) and human mammary carcinoma xenograft (B) samples using the following antibodies: rabbit anti-actin (clone 14-1) and rabbit anti-β1 integrin antibodies produced in our laboratory; rabbit anti-collagen I, mouse anti-GAPDH and rabbit anti-pan-histones antibodies were from Millipore. Following primary antibody incubation, the membranes were washed and incubated in the presence of HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibody from Jackson ImmunoResearch Laboratory. Finally, the membranes were washed and incubated in Western Lightning™ Chemiluminescence Reagent (PerkinElmer LAS). * indicates minimal residual histone contamination of the ECM-rich fraction. ** indicates partial depletion of monomeric (presumptively unassembled) collagen I in the CS fraction. *** indicates residual actin contamination of the ECM-rich fraction. The smear detected with the anti-collagen antibody represents different levels of post-translational modifications (e.g., cross-linking, glycosylation).
