Monitoring Activation of the Antiviral Pattern Recognition Receptors RIG-I And PKR By Limited Protease Digestion and Native PAGE
Özet
Innate defenses to virus infections are triggered by pattern recognition receptors (PRRs). The two cytoplasmic PRRs RIG-I and PKR bind to viral signature RNAs, change conformation, oligomerize, and activate antiviral signaling. Methods are described which allow to conveniently monitor the conformational switching and the oligomerization of these cytoplasmic PRRs.
Abstract
Host defenses to virus infection are dependent on a rapid detection by pattern recognition receptors (PRRs) of the innate immune system. In the cytoplasm, the PRRs RIG-I and PKR bind to specific viral RNA ligands. This first mediates conformational switching and oligomerization, and then enables activation of an antiviral interferon response. While methods to measure antiviral host gene expression are well established, methods to directly monitor the activation states of RIG-I and PKR are only partially and less well established.
Here, we describe two methods to monitor RIG-I and PKR stimulation upon infection with an established interferon inducer, the Rift Valley fever virus mutant clone 13 (Cl 13). Limited trypsin digestion allows to analyze alterations in protease sensitivity, indicating conformational changes of the PRRs. Trypsin digestion of lysates from mock infected cells results in a rapid degradation of RIG-I and PKR, whereas Cl 13 infection leads to the emergence of a protease-resistant RIG-I fragment. Also PKR shows a virus-induced partial resistance to trypsin digestion, which coincides with its hallmark phosphorylation at Thr 446. The formation of RIG-I and PKR oligomers was validated by native polyacrylamide gel electrophoresis (PAGE). Upon infection, there is a strong accumulation of RIG-I and PKR oligomeric complexes, whereas these proteins remained as monomers in mock infected samples.
Limited protease digestion and native PAGE, both coupled to western blot analysis, allow a sensitive and direct measurement of two diverse steps of RIG-I and PKR activation. These techniques are relatively easy and quick to perform and do not require expensive equipment.
Introduction
A crucial event in antiviral host defense is the rapid detection of the pathogen by the so-called pattern recognition receptors (PRRs)1,2. Intracellular detection of RNA virus infection is dependent on two cytoplasmic RNA helicases, RIG-I (retinoic acid inducible gene I) and MDA5 (melanoma differentiation associated protein 5)3-5. RIG-I is composed of two N-terminal caspase recruitment domains (CARDs), a central DECH-box type RNA helicase domain, and a C-terminal domain (CTD)4,6. Whereas the CTD and the helicase domain are required for recognition of non-self (viral) RNAs, the CARDs mediate downstream signaling leading to establishment of an antiviral host status.
If RIG-I is in the silent state, i.e. in the absence of a specific RNA ligand, the second CARD interacts with the central helicase domain and keeps RIG-I in an auto-inhibitory conformation7-11. RIG-I binds to short double-strand (ds) RNA bearing a 5’-triphosphate (5’PPP), long dsRNA, and polyU/UC-rich RNA, classic signature structures which are present on the genomes of many RNA viruses12-16. Two major characteristics of RIG-I activation are a switch to a closed conformation6,17 and the homo-oligomerization6,18,19. The conformational switch enhances RNA binding, exposes the CARDs for downstream signaling, and reconstitutes an active ATPase site8,9,11,20. The formation of oligomeric RIG-I complexes leads to enhanced recruitment of downstream signaling adaptor molecules to form a platform for antiviral signal transduction11. The RIG-I-regulated signaling chain eventually activates the transcription factor IRF-3 for up-regulation of interferon (IFN-alpha/beta) genes and hence the gene expression of interferon stimulated genes (ISGs) for a full antiviral response21,22. One of the best characterized ISGs is the RNA-activated protein kinase (PKR) 23. PKR belongs to the family of eukaryotic translation initiation factor 2 alpha (eIF2α) kinases and is composed of an N-terminal double-stranded RNA binding domain and a C-terminal kinase domain. The kinase domain constitutes the dimerization interface crucial for PKR activation and carries out the catalytic functions of the protein. Binding of PKR to viral dsRNA leads to its conformational change permitting dimerization and auto-phosphorylation at Thr 446 among other residues. PKR then mediates phosphorylation of eIF2α, thereby blocking the translation of viral mRNAs23-27.
Both RIG-I and PKR undergo major structural rearrangements, form oligomeric complexes and are post-translationally modified by phosphorylation/dephosphorylation and ubiquitination10,11,19,23,24,26-29. For a better understanding of which viral RNA structures are activating RIG-I and PKR (and at what stage viral antagonists could be interfering), it is important to precisely determine the activation status. For both PRRs it was previously described that activation leads to the emergence of trypsin-resistant protein fragments6,17,30 and higher-order oligomers6,18,19. However, given the wealth of literature on these key factors of the antiviral host response1,2,24, application of direct methods seems comparatively rare. In the hope of stimulating broader usage, we provide convenient and sensitive protocols to robustly analyze the activation states of RIG-I and PKR. The IFN competent human cell line A549 is infected with an established activator of RIG-I and PKR, the attenuated Rift Valley fever virus mutant clone 13 (Cl 13)31,32. After a simple lysing procedure, the extracts of infected cells are tested by limited trypsin digestion/western blot analysis to evaluate conformational switching, and by blue native polyacrylamide gel electrophoresis (PAGE) /Western blot analysis to measure formation of oligomers.
Protocol
1. Seeding of A549 Cells for Infection
- Cultivate a T75 flask of A549 cells at 37 °C and 5% CO2 in cell culture medium (DMEM supplemented with 10% FCS, 526.6 mg/l L-glutamine, 50.000 U/l penicillin, and 50 mg/l streptomycin).
- Before starting to harvest the cells, warm up cell culture medium, PBS and 0.05% trypsin-EDTA in a waterbath heated to 37 °C.
- Remove the medium and wash the cells with 10 ml PBS. Remove the PBS again.
- Add 3 ml of trypsin-EDTA and distribute equally in the flask. Transfer the flask in an incubator with 37 °C and 5% CO2.
- When all cells are detached, add 7 ml of cell culture medium, resuspend the cells, and transfer the cell suspension into a 15 ml Falcon tube.
- Centrifuge the cells at 800 x g for 5 min at RT, remove the supernatant and resuspend the pellet in 10 ml fresh cell culture medium.
- Count the cells with a counting chamber.
- Add 2.5 x 106 cells in 5 ml of cell culture medium in two T25 flasks each. Incubate for 16 hr at 37 °C and 5% CO2. One flask serves for the mock control and one for Cl 13 infection.
2. Infection with Rift Valley Fever Virus Clone 13 (Cl 13)
- NOTE: Cl 13 is an attenuated virus mutant which in Germany can be handled under BSL-2 conditions. Please refer to relevant national guidelines. Other typical IFN inducers would be Sendai virus (strain Cantell) or Newcastle disease virus.
- Pre-warm PBS, serum-free medium, and cell culture medium containing 5% FCS.
- Prepare 1.25 x 107 PFU/ml of Cl 13 in serum-free medium to infect 2.5 x 106 A549 cells with a multiplicity of infection (MOI) of 5. Prepare a slightly (roughly 10%) greater amount than needed to account for pipette errors.
- Wash the cells with PBS as described under 1.3.
- Add 1 ml of the Cl 13 dilution or of serum-free medium (uninfected control, mock) to the cells, and incubate for 1 hr at 37 °C and 5% CO2. Move the flask carefully every 15 min to ensure equal distribution of Cl 13 dilution and serum-free medium, respectively.
- After 1 hr of infection, remove the inocula, add 5 ml of pre-warmed cell culture medium with 5% FCS, and incubate for 5 hr at 37 °C and 5% CO2.
3. Preparation of Cell Lysates
- Prepare PBS / 0.5% Triton X-100 at 4 °C. Do not add serine protease inhibitors.
- Wash the cells with cold PBS and add 10 ml of fresh PBS.
- Scrape the cells off, transfer the cell suspension in a falcon tube, and centrifuge at 800 x g for 5 min at RT.
- Remove the supernatant and resuspend the cell pellet in 30 µl PBS / 0.5% Triton X-100. Transfer the lysate into a fresh 1.5 ml tube and incubate for at least 10 min at 4 °C.
- Centrifuge the lysate at 10.000 x g for 10 min at 4 °C and transfer the clarified cell lysate (supernatant) into a fresh tube.
- Determine the protein concentration by Bradford assay as described elsewhere33.
- Store at -20 °C or proceed to trypsin digestion (4.1) or native PAGE (5.1).
4. Determination of Conformational Changes of Pattern Recognition Receptors
- TPCK-trypsin Treatment of Cell Lysates
- Dilute L-1-tosylamido-2-phenylethyl chloromethyl ketone-treated (TPCK) trypsin in PBS to a final working concentration of 2 µg/µl.
- Adjust in two new tubes a final protein concentration of 25 µg of each protein lysate (mock or Cl 13) in a final volume of 9 µl with PBS. Hence, it should be four tubes with 25 µg lysate each, two times mock and two times Cl 13 infection. One set as input control (untreated) and one set for treatment with TPCK-trypsin.
- Add 1 µl of PBS (untreated) or 1 µl of 2 µg/µl TPCK-trypsin (final concentration: 0.2 µg/µl) to the cell lysates and mix the reactions by pipetting. DO NOT freeze and thaw TPCK-trypsin aliquots, because it will compromise the efficiency of digestion.
- Incubate the lysates at 37 °C for 25 min. Stop the reaction by adding 5x denaturing sample buffer (250 mM Tris-HCl pH 6.8, 10% SDS, 50% glycerol, 25% β-mercaptoethanol, 0.5% bromphenol blue) and by boiling for 5 min at 95 °C. It is important to NOT extend the trypsin incubation time. In case no protease-resistant fragments are detectable, the time of trypsin digestion must be shortened.
- After boiling, the samples can be stored at -20 °C.
- SDS Polyacrylamide Gel Electrophoresis (PAGE) and Western Blotting
- Load the samples on a sodium dodecyl sulfate (SDS) polyacrylamide gel containing a 5% stacking over a 12% resolving gel. Separate the proteins at 25 mA per gel until the bromphenol blue runs out.
- Activate a polyvinylidene fluoride (PVDF) membrane for 30 sec with methanol and put it into transfer buffer (48 mM Tris, 39 mM glycin, 1.3 mM SDS, 20% methanol).
- Prepare the blotting with a semidry blotting system and allow the transfer of the proteins at 10 V for 1 hr. Take the membrane out, rinse it briefly with water, and let it dry.
- Reactivate the membrane by shortly transferring into methanol. Wash for 5 min with TBS. Block with 10% skim milk in TBS for 1 hr at RT or at 4 °C O/N. Wash the membrane 3x for 5 min each with TBS.
- Prepare antibody dilution as recommended in table 1 and incubate the membrane for 1 hr at RT or at 4 °C O/N.
- Wash the membrane 3x for 10 min each with TBS-T. Add the appropriate secondary antibody coupled with horseradish peroxidase at a 1:20,000 dilution in 1% skim milk in TBS. Incubate 45 min at RT.
- Wash the membrane 3x for 10 min each with TBS-T, and one additional time with TBS.
- For signal detection use a commercial chemiluminescense kit and a digital gel imaging system.
- Coomassie Brilliant Blue G-250 Staining
- Perform SDS-PAGE as described in 4.2.1. Load samples on an SDS polyacrylamide gel and run the gel at 25 mA per gel until bromphenol blue runs out.
- Perform Coomassie Brilliant Blue G-250 staining at RT. Do all incubations under constant shaking.
- Transfer the gel to the fixation solution containing 40% methanol and 10% acetic acid for 30 min.
- Exchange the buffer to destaining solution (25% ethanol and 8% acetic acid) and incubate for 5 min.
- Stain the gel with 0.2% Coomassie Brillant Blue G-250 in 40% methanol and 10% acetic acid for 1 hr.
- Destain the gel with the destaining solution and exchange the buffer after 10 min, 30 min, and 60 min.
- Store the gel in 25% ethanol, 8% acetic acid, and 4% glycerol at 4 °C.
- Perform imaging and analysis as described under 4.2.8.
5. Analysis of Oligomeric States of Pattern Recognition Receptors
- Native PAGE
- Prepare 50 µg of cell lysate in a final volume of 10 µl with PBS and add 5× native sample buffer (250 mM Tris-HCl pH 6.8, 1% sodium deoxycholate, 50% glycerol, 0.5% bromphenol blue) to a final concentration of 1x.
- Load the samples IMMEDIATELY on a native polyacrylamide gel with 5% as stacking and 8% as resolving gel. Any delay will result in a loss of native complexes34.
- Run the gel at 20 mA per gel at 4 °C with 50 mM Tris-NaOH pH 9.0, 384 mM glycin as as anode and 50 mM Tris pH 8.3, 384 mM glycin, 1% sodium deoxycholate as cathode buffer. After 1.5-2 hr (bromphenol blue band has left the gel approximately 45 min earlier) the electrophoresis is finished.
- Western Blotting
- Activate a polyvinylidene fluoride (PVDF) membrane for 30 sec with methanol and put it into Towbin buffer (25 mM Tris, 192 mM glycin, 0.1% SDS, 20% methanol).
- Assemble a wet blot chamber according to the manufacturer’s instructions and fill the tank with Towin buffer.
- Perform the blotting with 250 mA for 1.5 hr at 4 °C.
- When blotting is finished proceed as described from 4.2.3 on.
Representative Results
Recognition of a viral agonist by RIG-I or PKR triggers conformational switching6,17,30 and oligomerization6,18,27. We assayed these two activation markers by limited protease digestion and native polyacrylamide gel electrophoresis (PAGE), respectively.
Human A549 cells were infected with Rift Valley fever virus clone 13 (Cl 13), which is characterized by a mutation of the IFN antagonist NSs35,36. Due to the absence of functional NSs, Clone 13 strongly induces RIG-I and PKR, leading to the establishment of a robust antiviral state in the cells12,31,32,37.
Trypsin digestion of mock infected cell lysates results in a rapid degradation of RIG-I, whereas Cl 13 infection leads to the generation of a 30 kDa resistant RIG-I fragment Figure 1A. Also PKR shows partial resistance to trypsin digestion in infected samples, which coincides with its phosphorylation Figure 1A. To monitor efficiency and specificity of trypsin digestion, gels were stained with Coomassie Brilliant Blue G-250. Untreated samples show equal amounts of loaded proteins Figure 1B. Subjecting mock and Cl 13 cell lysates to trypsin digestion leads to a comparable decrease of global protein amounts. This demonstrates that trypsin treatment has the same efficiency for mock and Cl 13-infected samples.
The formation of oligomeric complexes was assayed by native PAGE. In uninfected cells, only monomers of RIG-I and PKR were detected Figure 2. As additional control we included the transcription factor IRF-3, which is present as a monomer but known to dimerize upon activation via e.g., RIG-I38. Cl 13 infection leads to a strong accumulation of RIG-I oligomeric complexes in form of a smear and of PKR and IRF-3 dimers/oligomers as a defined protein band.
These results demonstrate that limited trypsin digestion and native PAGE are useful tools to monitor conformational changes and oligomer formation of RIG-I and PKR upon infection.
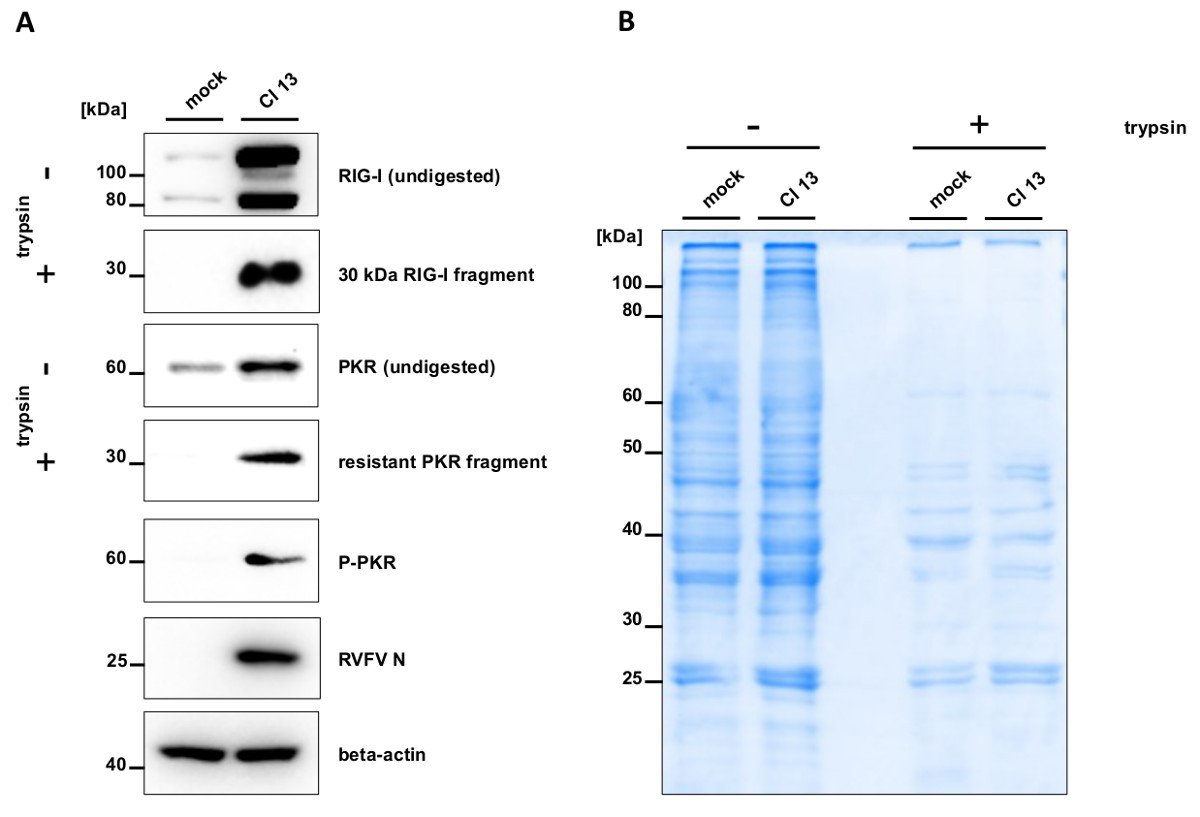
Figure 1. Conformational switch of RIG-I and PKR. A549 cells were mock infected or infected with Cl 13 at an MOI of 5. After 5 hr, cells were lysed in PBS supplemented with 0.5% Triton X-100 and cleared cell lysates were either left untreated or treated with trypsin. Samples were subjected to SDS PAGE followed by Western blotting (A) or Coomassie staining (B). Blots were stained against RIG-I, PKR, phosphorylated PKR (Thr 446) and against RVFV nucleoprotein (RVFV N) and beta-actin as infection and loading control, respectively. Please click here to view a larger version of this figure.
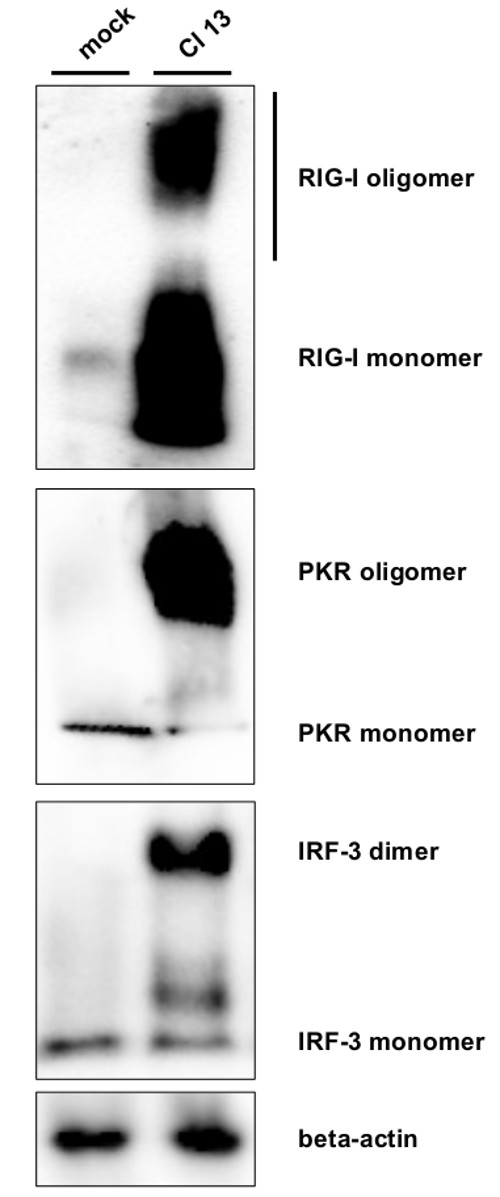
Figure 2. Oligomerization of RIG-I, PKR, and IRF-3. Cell lysates of mock and Cl 13-infected cell lysates were subjected to native PAGE followed by Western blot analysis. Staining was performed with antibodies against RIG-I, PKR, IRF-3, and beta-actin as loading control. PKR oligomers most likely represent dimers27. Please click here to view a larger version of this figure.
Discussion
Sensing the presence of viruses and activation of the antiviral type I IFN system are crucial for successful innate immune responses22. Virus detection is thereby mediated by pathogen recognition receptors (PRRs) like RIG-I and PKR, enabling a rapid response and activation of antiviral defense mechanisms. Here, we describe two methods to directly evaluate the activation status of RIG-I and PKR.
Limited protease digestion as a tool to monitor conformational changes of RIG-I and PKR was first described by the groups of M. Gale Jr. and T. Fujita6,17, and J. L. Cole30, respectively. It represents a sensitive method to evaluate sensitivity alterations to trypsin treatment caused by conformational changes. Applying trypsin digestion, we detected rapid degradation of RIG-I and PKR in mock infected samples, whereas trypsin treatment of virus-infected cell lysates led to trypsin resistant RIG-I fragments. Similarly, a trypsin resistant PKR fragment was detected upon Cl 13 infection. This was accompanied by PKR phosphorylation at Thr 446, a widely used marker of PKR activation. Comparing trypsin digestion of mock and Cl 13 cell lysates by Coomassie staining demonstrates a comparable decrease of global protein levels. This indicates that formation of resistant fragments is specific for proteins like RIG-I and PKR.
The formation of oligomeric complexes of RIG-I, PKR and IRF-3 was monitored by native PAGE. Subjecting Cl 13-infected cell lysates to native PAGE, we detected RIG-I, PKR and IRF-3 oligomers, whereas these proteins remained as monomers in mock infected samples. RIG-I needs to form oligomers to activate downstream pathways. It was hypothesized that RIG-I oligomerization supports recruitment of cofactors to form a signaling platform for antiviral response mechanisms11. The function of PKR dimerization is not entirely understood. Most likely, the PKR subunits in the dimer phosphorylate each other25. The type I IFN system is tightly regulated on a transcriptional level, and IRF-3 represents one central transcription factor for the induction of IFN and ISGs38. Central hallmarks of activation are phosphorylation, dimerization, and translocation to the nucleus, where it recruits the transcriptional co-activators p300 and CREB-binding protein (CBP) to initiate IFN mRNA synthesis21,39. Analysis of IRF-3 dimerization is a widely used tool to monitor activation of the type I IFN response38. Therefore, we used the detection of IRF-3 oligomeric complexes as a proof of principle for the oligomerization assays of RIG-I and PKR oligomerization. Indeed, allowing separation of protein lysates under non-denaturing conditions, we were able to detect IRF-3 dimerization in Cl 13 infected samples.
Undoubtedly, each lab will need to optimize the protocols of limited protease digestion and native PAGE. If confronted with no detection of any resistant proteins or too many resistant fragments, one might shorten or prolong the time of digestion, respectively. Differences can also be due to the specific TPCK-trypsin stock, as preparations slightly differ. Therefore, various TPCK-trypsin concentrations should be tested for optimization. Cell lysates can be prepared from other cell lines than A549, but adaptations of the protocol might be required. It is recommended to adjust the amount of total proteins for trypsin digestion and native PAGE according to the expression level of the protein of interest in this specific cell type. Moreover, limited detection or weak separation of oligomeric complexes by native PAGE can have several reasons and can be addressed as followed: make sure that the equipment is cleaned to remove remaining denaturing agents, keep all samples and the gel during the PAGE at 4 °C, and do not extend the time between sample preparation and loading onto the gel. Limited protease digestion and native PAGE have also been employed to monitor the activation of another important, closely related PRR, MDA540-42. Monitoring of MDA5 activation required different experimental conditions compared to RIG-I and PKR, and was therefore not included here.
In summary, point-by-point protocols for two useful and sensitive methods to measure activation of RIG-I and PKR are presented. Limited protease digestion and native PAGE, both coupled to Western blot analysis, permit monitoring of conformational changes and oligomerization, respectively. Using these methods, we had previously shown that RIG-I can be activated by nucleocapsids of various viruses directly after their entry into the cells32.
The exact nature and origin of the RNA species relevant for RIG-I and PKR activation in infected cells are still not entirely solved. Furthermore, many viruses interfere with the functions of PRRs by a wide variety of strategies12,43-46. The presented techniques enlarge the spectrum of methods by allowing an easy and direct measurement of RIG-I and PKR activation, are quick to perform, and do not require expensive equipment.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
We thank Alejandro Brun from CISA-INIA for providing anti-Rift Valley fever Virus sera. Work in our laboratories is supported by Forschungsförderung gem. §2 Abs. 3 Kooperationsvertrag Universitätsklinikum Giessen und Marburg, the Leibniz Graduate School for Emerging viral diseases (EIDIS), the DFG Sonderforschungsbereich (SFB) 1021, and the DFG Schwerpunktprogramm (SPP) 1596.
Materials
| Name | Company | Catalog Number | Yorumlar |
| cell culture | |||
| Dulbecco’s modified Eagle’s medium (DMEM) | Gibco | 21969-035 | |
| OptiMEM | Gibco | 31985-47 | |
| L-glutamine | PAA | 25030-024 | |
| penicillin-streptomycin | PAA | 15070-063 | |
| fetal calf serum (FCS) | PAA | 10270 | |
| 0.05% trypsin-EDTA | Gibco | 25300-054 | |
| chemicals | |||
| L-1-tosylamido-2-phenylethyl chloromethyl ketone-treated (TPCK) trypsin | Sigma Aldrich | T1426 | |
| antibodies | |||
| mouse monoclonal anti-RIG-I antibody (ALME-1) | Enzo Life Sciences | ALX-804-849-C100 | WB: 1:500 in 1% skim milk in TBS |
| mouse monoclonal anti-PKR (B10) | Santa Cruz | sc-6282 | WB: 1:500 in 1% skim milk in TBS |
| rabbit monoclonal anti-P-PKR (Thr446) | Epitomics | 1120-1 | WB: 1:1.000 in 5% BSA in TBS |
| rabbit polyclonal anti-IRF-3 | Santa Cruz | sc-9082 | WB: 1:500 in 1% skim milk in TBS |
| rabbit monoclonal anti-P-IRF-3 (Ser386) | IBL | og-413 | WB: 1:100 in 1% skim milk in TBS |
| rabbit anti-RVFV hyperimmune serum "C2" (MP-12) | Kindly provided by Alejandro Brun (CISA-INIA) | ||
| WB: 1:2.000 in 1% skim milk in TBS | |||
| mouse monoclonal anti-beta-actin (8H10D10) | Cell Signalling | 3700 | WB: 1:1.000 in 1% skim milk in TBS |
| polyclonal peroxidase-conjugated goat anti-rabbit | Thermo Fisher | 0031460 1892914 | WB: 1:20.000 in 1% skim milk in TBS |
| polyclonal peroxidase-conjugated goat anti-mouse | Thermo Fisher | 0031430 1892913 | WB: 1:20.000 in 1% skim milk in TBS |
Referanslar
- Gurtler, C., Bowie, A. G. Innate immune detection of microbial nucleic acids. Trends in Microbiology. 21, 413-420 (2013).
- Jensen, S., Thomsen, A. R. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. Journal of Virology. 86, 2900-2910 (2012).
- Kato, H., Takahasi, K., Fujita, T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunological Reviews. 243, 91-98 (2011).
- Yoneyama, M., et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nature Immunology. 5, 730-737 (2004).
- Andrejeva, J., et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proceedings of the National Academy of Sciences of the United States of America. 101, 17264-17269 (2004).
- Saito, T., et al. Regulation of innate antiviral defenses through a shared repressor domain. in RIG-I and LGP2 Proc Natl Acad Sci USA. 104, 582-587 (2007).
- Ferrage, F., et al. Structure and dynamics of the second CARD of human RIG-I provide mechanistic insights into regulation of RIG-I activation. Structure (London, England : 1993). 20, 2048-2061 (2012).
- Kolakofsky, D., Kowalinski, E., Cusack, S. . A structure-based model of RIG-I activation. 18, 2118-2127 (2012).
- Luo, D., Kohlway, A., Vela, A., Pyle, A. M. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure (London, England : 1993). 20, 1983-1988 (2012).
- Kowalinski, E., et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 147, 423-435 (2011).
- Luo, D., et al. Structural insights into RNA recognition by RIG-I. Cell. 147, 409-422 (2011).
- Habjan, M., et al. Processing of genome 5" termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PloS One. 3, e2032 (2008).
- Rehwinkel, J., Reis, E. S. C. Targeting the viral Achilles’ recognition of 5′-triphosphate RNA in innate anti-viral defence. Current Opinion in Microbiology. 16, 485-492 (2013).
- Pichlmair, A., et al. . RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. , 314-997 (2006).
- Hornung, V., et al. . 5′-Triphosphate RNA is the ligand for RIG-I. , 314-994 (2006).
- Saito, T., Gale, M. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. The Journal of Experimental Medicine. 205, 1523-1527 (2008).
- Takahasi, K., et al. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell. 29, 428-440 (2008).
- Binder, M., et al. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J Biol Chem. 286, 27278-27287 (2011).
- Jiang, X., et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 36, 959-973 (2012).
- Civril, F., et al. The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO reports. 12, 1127-1134 (2011).
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. The Journal of Biological Chemistry. 282, 15325-15329 (2007).
- Hertzog, P. J., Williams, B. R. Fine tuning type I interferon responses. Cytokin., & Growth Factor Reviews. 24, 217-225 (2013).
- Pindel, A., Sadler, A. The role of protein kinase R in the interferon response. Journal of Interfero., & Cytokine Research : the Official Journal of the International Society for Interferon and Cytokine Research. 31, 59-70 (2011).
- Donnelly, N., Gorman, A. M., Gupta, S., Samali, A. The eIF2alpha kinases: their structures and functions. Cellular and Molecular Life Sciences CMLS. 70, 3493-3511 (2013).
- Cole, J. L. Activation of PKR an open and shut case. Trends in Biochemical Sciences. 32, 57-62 (2007).
- Dauber, B., Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses. 1, 523-544 (2009).
- Dey, M., et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 122, 901-913 (2005).
- Gack, M. U., et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 446, 916-920 (2007).
- Zeng, W., et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 141, 315-330 (2010).
- Anderson, E., Cole, J. L. Domain stabilities in protein kinase R (PKR): evidence for weak interdomain interactions. Biyokimya. 47, 4887-4897 (2008).
- Habjan, M., et al. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. Journal of Virology. 83, 4365-4375 (2009).
- Weber, M., et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Hos., & Microbe. 13, 336-346 (2013).
- Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 72, 248-254 (1976).
- Wittig, I., Schagger, H. Advantages and limitations of clear-native. 5, 4338-4346 (2005).
- Muller, R., et al. Characterization of clone 13, a naturally attenuated avirulent isolate of Rift Valley fever virus, which is altered in the small segment. Am J Trop Med Hyg. 53, 405-411 (1995).
- Bouloy, M., et al. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. Journal of Virology. 75, 1371-1377 (2001).
- Ikegami, T., et al. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathogens. 5, e1000287 (2009).
- Iwamura, T., et al. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes to Cells : Devoted to Molecula., & Cellular Mechanisms. 6, 375-388 (2001).
- Yoneyama, M., Suhara, W., Fujita, T. Control of IRF-3 activation by phosphorylation. Journal of Interfero., & Cytokine Research : the Official Journal of the International Society for Interferon and Cytokine Research. 22, 73-76 (2002).
- Bamming, D., Horvath, C. M. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5 RIG-I and LGP2. The Journal of Biological Chemistry. 284, 9700-9712 (2009).
- Wu, B., et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 152, 276-289 (2013).
- Berke, I. C., Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. The EMBO Journal. 31, 1714-1726 (2012).
- Garcia-Sastre, A. Induction and evasion of type I interferon responses by influenza viruses. Virus Research. 162, 12-18 (2011).
- Taylor, K. E., Mossman, K. L. Recent advances in understanding viral evasion of type I interferon. İmmünoloji. 138, 190-197 (2013).
- Goodbourn, S., Randall, R. E. The regulation of type I interferon production by paramyxoviruses. Journal of Interfero., & Cytokine Research : the Official Journal of the International Society for Interferon and Cytokine Research. 29, 539-547 (2009).
- Versteeg, G. A., Garcia-Sastre, A. Viral tricks to grid-lock the type I interferon system. Current Opinion in Microbiology. 13, 508-516 (2010).
