One Mouse, Two Cultures: Isolation and Culture of Adult Neural Stem Cells from the Two Neurogenic Zones of Individual Mice
Özet
Here we describe a detailed protocol for the simultaneous generation of neural precursor cell cultures, as either adherent monolayers or neurospheres, from the subventricular zone and dentate gyrus of individual adult mice.
Abstract
The neurosphere assay and the adherent monolayer culture system are valuable tools to determine the potential (proliferation or differentiation) of adult neural stem cells in vitro. These assays can be used to compare the precursor potential of cells isolated from genetically different or differentially treated animals to determine the effects of exogenous factors on neural precursor cell proliferation and differentiation and to generate neural precursor cell lines that can be assayed over continuous passages. The neurosphere assay is traditionally used for the post-hoc identification of stem cells, primarily due to the lack of definitive markers with which they can be isolated from primary tissue and has the major advantage of giving a quick estimate of precursor cell numbers in brain tissue derived from individual animals. Adherent monolayer cultures, in contrast, are not traditionally used to compare proliferation between individual animals, as each culture is generally initiated from the combined tissue of between 5-8 animals. However, they have the major advantage that, unlike neurospheres, they consist of a mostly homogeneous population of precursor cells and are useful for following the differentiation process in single cells. Here, we describe, in detail, the generation of neurosphere cultures and, for the first time, adherent cultures from individual animals. This has many important implications including paired analysis of proliferation and/or differentiation potential in both the subventricular zone (SVZ) and dentate gyrus (DG) of treated or genetically different mouse lines, as well as a significant reduction in animal usage.
Introduction
The neurosphere assay1,2 and the adherent monolayer culture3,4, both developed in the early 1990s, still remain the gold standard in vitro neural stem cell assays. In these assays, primary tissue is micro dissected from a particular brain region, dissociated into a single cell suspension and cultured in the presence of the mitogens epidermal growth factor (EGF) and fibroblast growth factor-2 (FGF2) to form either free-floating clusters (neurospheres) or adherent monolayers. Both systems have a number of advantages and disadvantages and careful consideration should be given to the question that is to be addressed before one or the other system is chosen.
Neurospheres allow a straightforward read-out of differences in precursor cell number and potential. In addition, neurospheres are also a useful tool to study intrinsic specification of the cells when removed from their normal external environment. Extrinsic cues can be studied by simply adding the factor of interest to the growth medium and quantifying the number and size of the neurospheres generated. The major drawback of neurospheres however, is that they form their own niche, with the cells at the center of the neurospheres (particularly large neurospheres) being more differentiated than those on the surface5. Neurospheres contain a mix of stem cells, committed progenitors, and differentiated cells and the cell-cell interactions within the neurospheres counteract the maintenance of the stem cells. This is why neurospheres contain only a small number of true stem cells6-8.
Adherent monolayer cultures also provide a good in vitro system to model in vivo proliferation. Adherent cultures, in which the cells remain more isolated and homogeneous, can eliminate the heterogeneous nature of the neurosphere. Under these growth conditions the precursor cells proliferate rapidly and almost all cells are dividing and express the characteristic neural precursor markers Nestin, Sox2, and BLBP. The major disadvantage of the monolayer culture system compared to the neurosphere assay is that individual precursor-derived clones are unable to be monitored and quantified.
A drawback of most protocols for both types of cultures has been the necessity to use relatively large numbers of animals, because the yield of the isolation strategies has often been poor. At the same time, it has become clear that adult neurogenesis contributes to the individualization of the brain9, resulting in the need for individualized ex vivo models as well. These needs can be met by "one-mouse-one-culture" protocols as described in this report.
The following visual protocol describes the simultaneous generation of neural precursor cultures from both the SVZ and DG of individual animals either as adherent monolayers or as neurospheres. The generation of cultures from individual animals is particularly useful when comparisons between individually treated animals or various individual transgenic or wild-type mice are required. This protocol includes detailed instructions for the simultaneous microdissection of the SVZ and DG regions from adult mice, their dissociation into a single cell suspension, in vitro culture as either adherent monolayer cultures or neurospheres and analysis of multipotentiality and long-term potential, the two cardinal properties of a bone fide stem cell.
Protocol
1. Basic Setup and Preparation of Culture Medium
- At least two days prior to commencing the experiment, prepare Poly-D-lysine (PDL)/Laminin coated plates for adherent monolayer cultures. To prepare wells/flasks add enough PDL (10 µg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Allow to air dry. Add Laminin (5 µg/ml in cold DMEM:F12) and incubate at 37 °C overnight. Remove the Laminin and either use the plates immediately or store with the Laminin at -20 °C until required.
- Prepare fire polished pipettes with "medium" and "small" bores by rotating glass Pasteur pipettes in a flame until the edges become rounded. Autoclave to sterilize.
- On the day of dissection, prepare the appropriate amount of culture medium by mixing Neural Basal Medium with 2% B27, 1x GlutaMAX, 2 µg/ml heparin, 50 units/ml Penicillin/Streptomycin, 20 ng/ml purified mouse receptor-grade epidermal growth factor (EGF), and 20 ng/ml recombinant bovine fibroblast growth factor (FGF-2). Warm the culture medium to 37 °C in a water bath.
- For the SVZ dissociation, prepare 0.05% Trypsin-EDTA and 0.125 mg/ml Trypsin inhibitor containing 0.01 mg/ml DNaseI. Equilibrate these solutions to 37 °C.
- Set up a dissection microscope and prepare the tools needed to remove the brain (scissors and small spatula) and for SVZ and DG dissections (scalpel, 27 G needle attached to 1 ml syringe, 1 x #7 forceps, 1 x #5/45 forceps) by soaking in 70% ethanol.
2. Harvesting of Adult Mouse Brains and SVZ/DG Microdissections
- Anesthetize single adult (8-week-old) mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- Spray the head with 70% ethanol to sterilize the area and to minimize the amount of fur that adheres to the scissors and brain. Using sharp scissors decapitate the animal at the base of the brain stem.
- Holding the head at the base of the skull, cut the skull between the two olfactory bulbs by placing one blade of a small pair of scissors into each eye cavity and cutting coronally. Next, make two lateral cuts at the base of the skull, followed by a longitudinal cut through the skull along the sagittal suture. Caution: ensure the angle of the scissors is as shallow as possible to avoid damaging the underlying brain.
- Expose the brain by peeling back the skull with either the blade of the scissors or a pair of curved forceps. Free the brain from the skull using a small spatula and place into cold PBS.
- Rinse brains with PBS to remove blood and fur.
- Transfer brains to a 10 cm plastic Petri dish containing PBS
- Place Petri dish containing the brain under a dissecting microscope at low magnification and position the brain on its ventral surface. Using fine curved forceps remove the olfactory bulbs while holding the brain in position by the cerebellum.
- Rotate the brain onto the dorsal aspect and using a scalpel make a coronal cut through the brain at the level of the optic chiasm
- To microdissect the SVZ (for more detailed instructions, see also Azari et al.10), place the rostral portion of the brain so that the cut coronal surface faces upwards and focus the microscope onto a higher magnification. Remove and discard the septum using fine curved forceps.
- Dissect the SVZ (the thin layer of tissue surrounding the ventricle) by placing the tip of one blade of a pair of fine curved forceps in the lateral corner of the lateral ventricle immediately under the corpus callosum and the other approximately 1 mm into the tissue immediately adjacent to the ventricle. Press down the forceps towards the base of the dish and towards the ventral aspect of the ventricle to remove a small triangular piece of tissue. Place the dissected SVZ into a Petri dish on ice.
- To microdissect the DG (for more detailed instructions, see also Hagihara et al.11), place the caudal portion of the brain in the Petri dish and cut along the longitudinal fissure using a scalpel.
- Under a dissection microscope, remove the cerebellum and the diencephalon using forceps.
- Refocus the microscope so that the borders around the DG are now visible. To remove the dentate gyrus, insert the tip of a 27 G needle and slide along the border between the DG and Ammon's horn. Using the fine forceps, free the DG from the surrounding tissue.
3. SVZ Tissue Dissociation
- Mince the tissue using a scalpel blade for approximately 1 min until no large pieces remain.
- Transfer the minced tissue to a 15 ml tube using 1 ml of prewarmed 0.05% Trypsin-EDTA and incubate for 7 min in a water bath set to 37 °C.
- To stop the enzymatic reaction, add 1 ml of trypsin inhibitor containing DNaseI and mix the contents by flicking the tube.
- Pellet the suspension by centrifugation at 300 x g for 5 min and discard the supernatant
- Resuspend the pellet in 1 ml of growth medium and dissociate by gently pipetting up and down approximately 7-10x using a P1000 pipette. Caution: over triturating can lead to increased cell death and will negatively impact on subsequent cell growth.
- Add growth medium to a total volume of 5 ml and pass the cell suspension through a 40 µm sieve to remove debris and undissociated tissue clumps.
- Centrifuge at 300 x g for 5 min, discard the supernatant and resuspend the resulting pellet in 200 µl growth medium.
4. DG Tissue Dissociation
- Mince the tissue using a scalpel blade for approximately 1 min until no large pieces remain and transfer into prewarmed PDD enzyme mix (Papain 2.5 U/ml, Dispase 1 U/ml, DNaseI 250 U/ml). Incubate for 20 min at 37 °C, mixing by inverting the tube every 3-5 min.
- Dissociate the tissue mechanically using a medium bore, fire polished, Pasteur pipette by pipetting up and down gently 10x.
- Incubate for a further 10 min at 37 °C, mixing by inverting the tube every 3-5 min.
- Further dissociate the tissue mechanically using a small bore, fire polished Pasteur pipette by pipetting up and down gently 10x.
- Centrifuge at 130 x g for 5 min.
- Remove the supernatant and resuspend the pellet in 1 ml buffer solution (1x HBSS, 30 mM Glucose, 2 mM HEPES (pH 7.4), 26 mM NaHCO3). Make up to 10 ml with buffer solution.
- Centrifuge at 130 x g for 5 min.
- Remove supernatant and resuspend the pellet in 5 ml of 20% Percoll. (To prepare 90% Percoll, add 4.5 ml of 100% Percoll to 0.5 ml of 10x PBS then further dilute this to 20% by adding 1.1 ml 90% Percoll to 3.9 ml 1x PBS).
- Centrifuge 450 x g for 15 min.
- Remove the supernatant and resuspend the pellet in 10 ml buffer.
- Centrifuge at 130 x g for 5 min.
- Resuspend the pellet in 200 µl growth medium.
5. Generation of Adherent Monolayer Cultures
- Plate the dissociated SVZ or DG tissue into a single PDL/Laminin coated well of a 96-well plate and incubate at 37 °C with 5% CO2.
- Approximately 24 hr after plating, once the cells have adhered to the coated surface, exchange the growth medium to further remove excess debris.
- Every subsequent 3-4 days, exchange half of the growth medium with fresh medium to replenish the growth factors.
- Repeat until the cells reach approximately 80% confluency and are ready to be passaged. Note: the time between plating and the first passage can take up to 2-3 weeks.
6. Passaging of Adherent Monolayer Cultures
- When the cultures reach approximately 80% confluency remove the medium from the well and wash with PBS.
Note: Do not allow the cells to exceed 90% confluency as this can lead to detachment of cells and neurosphere formation and in addition, increased levels of cell death. - Add 50 µl Accutase and incubate at 37 °C for 2-3 min (checking to see if the cells are rounded and detached).
- Remove the cells to a 15 ml tube and wash the well once with PBS and transfer to the same tube.
- Dilute cells to 5 ml with PBS and centrifuge 300 x g for 5 min.
- For the first passage, dilute cells to 1 ml and plate into a PDL/Laminin coated well of a 24 well plate.
- For subsequent passages, resuspend cells in 200 µl growth medium and count using a hemocytometer. Plate at 1 x 104 cells/cm2 in the appropriate sized coated well or flask.
7. Differentiation of Adherent Monolayer Cultures
- To differentiate the adherent monolayer cultures, plate proliferating cells onto PDL/Laminin coated coverslips at a density of 1 x 104 cells/cm2 in growth medium containing 20 ng/ml EGF and 10 ng/ml bFGF.
- When the cells reach approximately 80% confluency (usually 2 days), replace the growth medium with medium containing 5 ng/ml bFGF and 0 ng/ml EGF.
- Following 2 days in 5 ng/ml bFGF, replace the medium with growth medium in the absence of both mitogens for a further 3 days. Note: during this period a significant amount of cell death will occur.
- After a total of 5 days, wash the differentiated cells with PBS to remove any dead cells then fix with 4% paraformaldehyde (PFA) for 20 min at room temperature.
- Wash again with PBS to remove any PFA and store coverslips in wells in 1 ml PBS at 4 °C.
8. Neurosphere Culture and Quantification
- Dilute the dissociated SVZ or DG tissue from one animal in 20 ml of culture medium and plate 200 µl/well across a 96-well plate using a 10 ml multidoser pipette.
- Incubate at 37 °C with 5% CO2 for 6-7 days for SVZ-derived neurospheres and 10-12 days for DG-derived neurospheres.
Note: growth for longer than these recommended incubation times will result in overgrowth and will lead to cell death in the center of the neurospheres and/or, spontaneous attachment and differentiation. - Count and measure the diameter of the neurospheres using an eyepiece graticule fitted to an upright light microscope
9. Passaging the Neurospheres
After the primary neurospheres have been counted and their size recorded they can be expanded over several passages beginning with either a single neurosphere or a bulk culture.
- Bulk culture neurosphere expansion
- To passage combined neurospheres as a bulk culture, remove the medium containing the neurospheres from the plate, transfer to a 15 ml tube and centrifuge at 300 x g for 5 min.
- Discard the supernatant and resuspend the neurospheres in 1 ml of prewarmed 0.05% Trypsin-EDTA and incubate at room temperature for 3 min.
- Add an equal volume of trypsin inhibitor containing DNaseI and mix well.
- Centrifuge for 5 min at 300 x g, remove the supernatant and add 1 ml of growth medium.
- Triturate up and down approximately 10x with a P1000 pipette to dissociate the neurospheres.
- Remove 10 µl of the cell suspension and mix with an equal volume of trypan blue and perform a live cell count using a hemocytometer.
- Reseed the cells at a density of 1 x 104 cells/cm2 in the appropriate sized cell culture well or flask.
- Incubate at 37 °C with 5% CO2 until secondary neurospheres form.
- Single neurosphere expansion
- To passage individual neurospheres choose wells that contain a single neurosphere and carefully remove and discard approximately 160 µl of the growth medium from each well without disturbing the neurosphere.
- Add 100 µl of 0.05% Trypsin-EDTA to each well to be passaged and incubate at room temperature for 3 min.
- Add 100 µl of Trypsin inhibitor containing DNAseI to stop the reaction.
- Triturate approximately 10 times up and down with a P200 pipette to break apart the neurosphere.
- Transfer the 200 µl containing the dissociated neurosphere to a new well of a 24-well plate containing 1.5 ml of growth medium. Incubate at 37 °C with 5% CO2 until secondary neurospheres form.
Note: to determine long-term potential, one of the cardinal properties of a true neural stem cell, neurospheres should be passaged for at least 5-10 passages. See also the literature on the in part controversial interpretation of such results12-14.
10. Differentiation of Neurosphere Cultures
Primary or passaged neurospheres can be differentiated to determine multipotentiality.
- Remove neurospheres in suspension from their culture plate or flask and transfer them to a 10 cm plastic Petri dish.
- Under a dissection microscope remove approximately 15-20 neurospheres from the medium using a P20 pipette and transfer to a 24-well plate containing the culture medium without growth factors and a PDL/Laminin coated coverslip.
- Differentiate for approximately 7 days at 37 °C with 5% CO2.
- Wash the differentiated neurospheres with PBS to remove any dead cells then fix with 4% PFA for 20 min at room temperature.
- Wash again with PBS to remove any PFA and store coverslips in wells in 1 ml PBS at 4 °C
11. Immunostaining of Neurosphere and Adherent Cultures
Note: For staining with the O4 antibody omit the Triton from the blocking and staining steps and remember to use an appropriate IgM secondary antibody.
- Incubate the coverslips containing the differentiated neurospheres or adherent monolayer cultures in blocking solution (10% Normal Donkey Serum in PBS containing 0.2% Triton X-100) for 60 min at room temperature.
- Incubate in fresh blocking solution containing primary bIII-tubulin, Map2a+b, glial fibrillary acidic protein (GFAP) or O4 antibodies for 60min at room temperature.
- Wash 3x with PBS.
- Incubate in fresh blocking solution containing appropriate fluorescence conjugated secondary antibodies and 4,6-diamidino-2-phenylindole (DAPI; 1:5,000) for 30 min at room temperature in the dark.
- Wash three times with PBS.
- Mount the coverslips onto microscope slides using fluorescence mounting medium and air dry in the dark overnight
- View and image using a fluorescence microscope.
Representative Results
Although the two neurogenic regions of the adult mouse brain both contain neural precursor cells, these cells can behave quite differently when cultured in vitro. The adherent monolayer cultures generated from both regions appear morphologically indistinguishable (Figure 1A), however, the SVZ-derived adherent cultures proliferate faster and need to be passaged, on average, 1-2 days earlier than those derived from the DG. As neurospheres, the SVZ-derived precursor cells also proliferate faster and form larger neurospheres (Figure 1B) than the DG-derived precursor cells (Figure 1C). Whilst SVZ-derived neurospheres are typically counted after 6-7 days in culture, DG-derived neurospheres are usually quantified after 10-12 days. In addition, a far greater number of neural precursor cells reside in the SVZ compared to the DG, as evidenced by the almost 10-fold greater number of neurospheres that can be generated from this region (SVZ: 1,173±74.9 vs. DG: 145.3±26.4; p = <0.0001; n = 10 animals per group; Figure 2A).
Studies have shown that the precursor cells within the SVZ and DG respond to different stimuli. The precursor cells in the DG are activated by specific types of spatial learning and by stimuli such as environmental enrichment and physical activity, whereas the SVZ precursor cells are activated by olfactory learning and olfactory enrichment. Consistent with this, one of us (TLW) previously demonstrated that the DG contains a population of latent stem and progenitor cells that can be activated by neural excitation15-18. In contrast, we found that the SVZ precursor cells respond quite differently to this stimulus, with a decrease in neurosphere number in response to depolarizing levels of KCl 17. Here, we have repeated this experiment, plating half of the isolated cells derived from the SVZ and DG of individual animals in depolarizing levels of KCl and the other half in control KCl levels. We demonstrate, as previously, that while the DG precursor cells are activated by depolarization (101.2±17.4 vs. 184.8±12.5, p = 0.005, n = 5 animals), the proliferation of the SVZ-derived cells is in fact significantly decreased (368.0±62.9 vs 266.6±41.6, p = 0.02, n = 5 animals; Figure 2B).
To confirm long-term potential, one of the cardinal features of a true stem cell, single neurospheres or adherent monolayer cultures must be capable of extended expansion i.e. over at least 10 passages. At each passage, following the preparation of a single-cell suspension, the number of cells is counted and the fold expansion is calculated. The theoretical cell total is then calculated by multiplying the fold expansion during that passage by the theoretical total from the previous passage. This is displayed as a line graph with the passage number plotted against the log10 of the theoretical total cell number (see example Figure 3). To confirm multipotentiality, both monolayer cultures and neurospheres can be differentiated by mitogen withdrawal and be shown to give rise to both neurons, and glia (Figure 4).
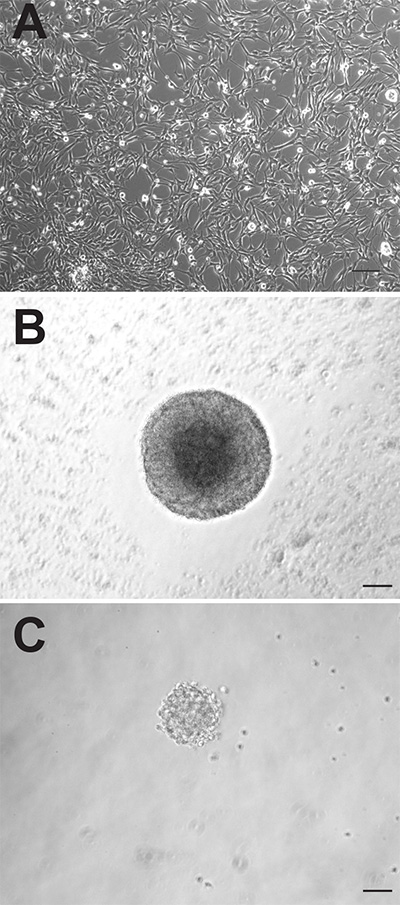
Figure 1. Adult mouse precursor cells can be cultured as adherent monolayer cultures (A) or as neurospheres (B: SVZ, C: DG). Scale bar is 50 µm. Click here to view larger image.
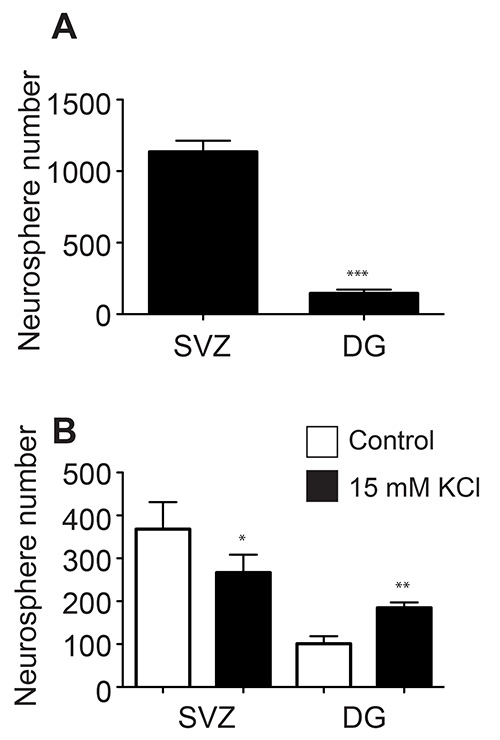
Figure 2. Significantly more neurospheres are generated from the SVZ compared to the DG of single mice (A). The SVZ and DG precursor cells respond differently to in vitro depolarization (B).
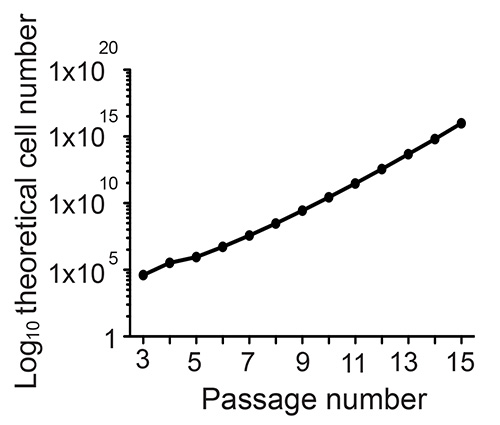
Figure 3. To confirm long-term potentiation, neurospheres are expanded for over 10 passages.
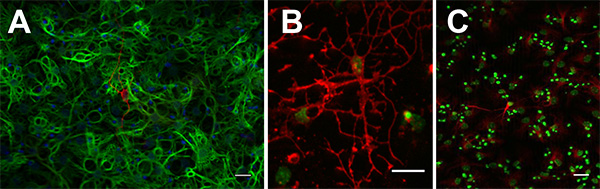
Figure 4. Neurospheres can be differentiated into bIII-tubulin+ neurons (A: red), GFAP+ astrocytes (A: green), O4+ oligodendrocytes (B: red) and Map2ab+ neurons (C: red). Click here to view larger image.
Discussion
This paper presents a detailed protocol for the initiation of neural precursor cultures, both as adherent monolayers and neurospheres, from the two major neurogenic regions of the adult mouse brain. There are a number of important points that must be kept in mind when attempting either of these in vitro culture systems. Firstly, the choice of dissociation method is very important and is tissue dependent. In our hands, 0.05% trypsin-EDTA is very effective for dissociation of SVZ tissue, and results in a higher number of neurospheres than when using a papain-based dissociation technique. For the dissociation of DG tissue however, we strongly recommend a papain-based dissociation approach. When directly comparing the two dissociation methods on DG tissue, we observed a significantly lower yield of viable cells and approximately 10-fold fewer neurospheres when using trypsin. This difference in dissociation could be due to the difference in tissue composition between the two regions. The compact tissue of the DG is surrounded by extensive neuropil and extensive damage of cellular processes can occur during dissociation.
A second important point to note is that, while the neurosphere assay can be useful to make quantitative statements about the number of precursor cells present in a given tissue sample, some caution must, however, be employed in the interpretation of these absolute numbers. Fusion of neurospheres can be a major confounding factor. Several studies have shown that neurons are highly motile and can fuse, even under what are supposedly ‘clonal’ conditions7,19. The resulting neurosphere frequency can be very dependent on factors including the medium components, the dissection procedure and the dissociation process. Even between experienced handlers some variation in the number of neurospheres generated from supposedly identical samples is evident (see Figure 1A.) More useful, is a direct comparison of the precursor frequency between two given samples (i.e. control vs. treated or wild-type vs. knock-out) handled by the same person within a single experiment, rather than a quantitative statement of total precursor cell number.
When deciding which of the two culture methods is most suited for a particular experiment it is important to note that these two culture systems differ in the homogeneity of the cell types generated. In comparison to proliferating adherent cell cultures, which show a fairly homogeneous precursor cell pool (~98% of cells are Sox2+)20, neurospheres are more heterogeneous and contain, as well as proliferating precursor cells, differentiated neurons, and astrocytes21,22. It is important that the neurospheres are not cultured for extended periods between passages as the larger the neurosphere become the more likely it is to find differentiated cell types in their core.
We traditionally initiate adherent monolayer neural precursor cultures from the DG tissue of between 5-8 mice. Therefore, when attempting to establish the adherent monolayer cultures from the DG or SVZ of a single mouse, the utmost care needs to be taken during the tissue dissociation procedure in order to avoid excessive cell death caused by over triturating of the tissue, or taking extended periods of time between the dissection and final culturing steps. This protocol describes, for the first time, the generation of adherent monolayer precursor cultures from both the SVZ and DG of individual animals. There are many instances when the comparison of precursor proliferation and differentiation needs to be made on a single animal basis. These include the ability to directly compare the DG and SVZ of individual animals using paired statistics and to pair culture data with individual behavioral or physiological data9. Single animal cultures also allow the use of rare transgenic animals, where age-matching a pool of 5-8 donors per culture is not possible, as well as unique animals (e.g. F2 crosses or out bred animals) for genetic association studies.
Açıklamalar
The authors have nothing to disclose.
Acknowledgements
TLW was supported by a Marie Curie International Incoming Fellowship. This work was also financed from basic institutional funding, Bundesministerium für Bildung and Forschung (BMBF) funding and partly with support from Priority Research Program (SFB) 655 to GK. The authors would like to thank Anne Karasinsky for care and maintenance of all animals used in this study and Odette Leiter, Susann Ruhwald, Fanny Boehme, and Richard Wetzel for cell culture and microscopy assistance.
Materials
| CONSUMABLES | |||
| poly-D-lysine | Sigma | P7280-5MG | |
| laminin | Roche | 11243217001 | |
| glass pastuer pipettes | Volac | BS5732 | |
| DMEM:F12 (1:1) 1X | Life Technologies | 21331-020 | |
| Neural Basal Medium (1X) | Life Technologies | 21103-049 | |
| B27 supplements (50X) | Life Technologies | 17504-044 | |
| Glutamax | Life Technologies | 35050-038 | |
| Heparain | Sigma | H3393 | |
| Penacillin/Streptomycin | Life Technologies | 15140-122 | |
| EGF | PeproTech | AF-100-15 | |
| bFGF | PeproTech | 100-18B | |
| 0.05% trypsin-EDTA | Life Technologies | 25300-054 | |
| trypsin inhibitor | Sigma | T6522 | |
| DNaseI | Roche | 10104159001 | |
| Accutase | PAA | L11-007 | |
| Papain | Worthington | LS003120 | |
| Dispase | Life Technologies | 17105-041 | |
| Percoll | GE Healthcare | 17-0891-02 | |
| HBSS (with Calcium & Magnesium) | Life Technologies | 14025-050 | |
| Glucose | Roth | X997.2 | |
| HEPES | Sigma | H3375-500G | |
| NaHCO3 | Merck | K39347429847 | |
| 1mL syringes | Braun | 2016-10 | |
| 27 Gauge needles | Braun | 4657705 | |
| scalpels (#22 disposable) | Braun | BA222 | |
| Dumont #7 forceps | FST | 11271-30 | |
| Dumont 5/45 forceps | FST | 11251-35 | |
| scissors | FST | 14060-10 | |
| Iris spatula | FST | 10093-13 | |
| 70% ethanol | |||
| PBS | Life Technologies | 14040-091 | |
| flasks/well plates | TPP | 92696 | |
| PFA (4%) | Sigma | P6148 | |
| hemocytometer | Marienfeld | 650010 | |
| trypan blue (0.4%) | Sigma | T8154 | |
| NDS | Millipore | 530 | |
| TritonX-100 | Sigma | T9284 | |
| mouse monoclonal bIII-tubulin antibody | Promega | G712A | |
| rabbit polyclonal glial fibrillary acidic protein antibody | Dako | 20334 | |
| O4 | R & D systems | MAB1326 | |
| Map2a+b | Sigma | M1406 | |
| donkey anti-mouse Cy3 antibody | Jackson ImmunoResearch | 715-505-151 | |
| donkey anti-rabbit Alexa488 | Dianova | 711-545-152 | |
| 4,6-diamidino-2-phenylindole (DAPI) | Invitrogen | 861405 | |
| Aqua Polymount | Polysciences Inc | 18606 | |
| 10 ml Combi tips | eppendorf | 30089677 | |
| plastic 10ml and 25mL serological pipettes | Corning | 4488/4489 | |
| EQUIPMENT | |||
| Pipetboy | Integra biosciences | 521942 | |
| multidoser pipette | eppendorf | ||
| 37C waterbath | |||
| dissecting microscope | |||
| 37C:5%Co2 incubator | |||
| centrifuge | eppendorf | 5810R |
Referanslar
- Reynolds, B. A., Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 255, 1707-1710 (1992).
- Reynolds, B. A., Weiss, S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 175, 1-13 (1996).
- Palmer, T. D., Ray, J., Gage, F. H. FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol. Cell. Neurosci. 6, 474-486 (1995).
- Ray, J., Raymon, H. K., Gage, F. H. Generation and culturing of precursor cells and neuroblasts from embryonic and adult central nervous system. Methods Enzymol. 254, 20-37 (1995).
- Bez, A., et al. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 993, 18-29 (2003).
- Babu, H., Cheung, G., Kettenmann, H., Palmer, T. D., Kempermann, G. Enriched monolayer precursor cell cultures from micro-dissected adult mouse dentate gyrus yield functional granule cell-like neurons. PLoS One. 2, (2007).
- Jessberger, S., Clemenson, G. D., Gage, F. H. Spontaneous fusion and nonclonal growth of adult neural stem cells. Stem Cells. 25, 871-874 (2007).
- Reynolds, B. A., Tetzlaff, W., Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 12, 4565-4574 (1992).
- Freund, J., et al. Emergence of individuality in genetically identical mice. Science. 340, 756-759 (2013).
- Azari, H., Rahman, M., Sharififar, S., Reynolds, B. A. Isolation and expansion of the adult mouse neural stem cells using the neurosphere assay. J. Vis. Exp. , (2010).
- Hagihara, H., Toyama, K., Yamasaki, N., Miyakawa, T. Dissection of hippocampal dentate gyrus from adult mouse. J. Vis. Exp. , (2009).
- Jensen, J. B., Parmar, M. Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 34, 153-161 (2006).
- Pastrana, E., Silva-Vargas, V., Doetsch, F. Eyes wide open: a critical review of sphere-formation as an assay for stem cells. Cell. Stem Cell. 8, 486-498 (2011).
- Reynolds, B. A., Rietze, R. L. Neural stem cells and neurospheres- re-evaluating the relationship. Nat. Methods. 2, 333-336 (2005).
- Walker, T. L., Turnbull, G. W., Mackay, E. W., Hannan, A. J., Bartlett, P. F. The latent stem cell population is retained in the hippocampus of transgenic Huntington’s disease mice but not wild-type mice. PLoS One. 6, (2011).
- Walker, T. L., et al. Prolactin stimulates precursor cells in the adult mouse hippocampus. PLoS One. 7, (2012).
- Walker, T. L., et al. Latent stem and progenitor cells in the hippocampus are activated by neural excitation. J. Neurosci. 28, 5240-5247 (2008).
- Walker, T. L., et al. Prominin-1 allows prospective isolation of neural stem cells from the adult murine hippocampus. J. Neurosci. 33, (2013).
- Singec, I., et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods. 3, 801-806 (2006).
- Babu, H., et al. A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front. Neurosci. 5, 89 (2011).
- Parmar, M., Sjoberg, A., Bjorklund, A., Kokaia, Z. Phenotypic and molecular identity of cells in the adult subventricular zone in vivo and after expansion in vitro. Mol. Cell Neurosci. 24, 741-752 (2003).
- Suslov, O. N., Kukekov, V. G., Ignatova, T. N., Steindler, D. A. Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc. Natl. Acad. Sci. U.S.A. 99, 14506-14511 (2002).
