Representative results of the three examples are shown in Figures 4 to 6. The first approach aimed at deleting the neighboring genes ctxA and ctxB. Together they encode the major virulence factor of V. cholerae, cholera toxin. We designed oligonucleotides for the TransFLP method as described above and according to Figure 3. The parental strain, the intermediate strain harboring the FRT-flanked antibiotic resistance cassette at the original DNA locus of ctxAB and the final strain deleted for ctxAB and freed of the resistance cassette were all tested for their genotype (Figure 4). The method of choice was whole-cell PCR. We used primers that do not anneal to the transforming PCR fragment but only to chromosomal DNA (Figure 3). This allowed us to verify the site-specific exchange of the PCR fragment with the homologous region of the chromosome (Figure 4A). The PCR fragments were separated by standard agarose gel electrophoresis to determine their size (Figure 4B). This strategy can be used to check strain intermediates and to confirm the final strain construct(s).
For the second example, the removal of a whole genomic island, we chose the Vibrio pathogenicity island 1 (VPI-1) as target13. The standard TransFLP procedure described above was unsuccessful in this case. It was known from previous studies that such a large DNA region can be exchanged by a similar sized DNA region using chitin-induced natural transformation9. We therefore hypothesized that the size difference between the region-to-be-deleted and the rather short transforming PCR fragment (< 3.5 kb) might be the limiting factor. We therefore decided to use the TransFLP method in two steps: we first integrated the aph gene (kanR) preceded by a single FRT site at the start of VPI-1 (illustrated in Figure 5A). We then performed a second round of natural transformation, which allowed us to insert an additional antibiotic resistance cassette (gmR, conferring gentamicin resistance) followed by a second FRT site at the end of the pathogenicity island (Figure 5A). The respective double-resistant strain was electroporated to insert plasmid pBR-flp and the TransFLP method was continued as described above. The resulting strain lacked the whole VPI-1 as verified by PCR of purified genomic DNA (Figure 5B).
For the third example we integrated a T7 RNA polymerase-dependent promoter sequence upstream of a gene-of-interest, in our case the gene encoding the major regulator of natural transformation tfoX6. This was done using the TransFLP method with a slight modification from the standard protocol: we included the T7 RNA dependent promoter consensus sequence according to Ref.14 as overhangs into the oligonucleotides #4 and #5. With this strategy the T7 RNA polymerase-dependent promoter sequence was kept on the chromosome even after the antibiotic resistance cassette was excised (Figure 6). We integrated the construct into V. cholerae strain ADVCH-T7POL, which harbors the T7 RNA polymerase gene driven by the lacUV5 promoter (Blokesch, unpublished). To test for the functionality of the construct, namely the T7 RNA polymerase-dependent expression of tfoX, we performed natural transformation assays in LB medium. The parental strain is not naturally transformable under these conditions due to the lack of tfoX transcription in rich medium and in the absence of its natural inducer chitin (Figure 6). This phenotype was independent of whether the T7 RNA polymerase was artificially induced by IPTG or not (Figure 6, lanes 1 and 2, respectively). However, the TransFLP-generated strain ADVCH-T7POL-T7-tfoX::FRT, which contains the T7 RNA polymerase-dependent promoter upstream of the competence gene tfoX, was indeed naturally transformable in rich medium (Figure 6, lanes 3 and 4). Due to the leakage of the lacUV5 promoter in LB medium15 the produced T7 RNA polymerase already transcribed tfoX from the T7 RNA polymerase-dependent promoter without induction. This initiated natural competence and transformation confirming the functionality of the integrated T7 RNA polymerase-dependent promoter sequence (Figure 6, lane 3). This phenotype was significantly enhanced by full induction of the T7 RNA polymerase due to addition of the inducer of the lacUV5 promoter, IPTG (Figure 6, lane 4).
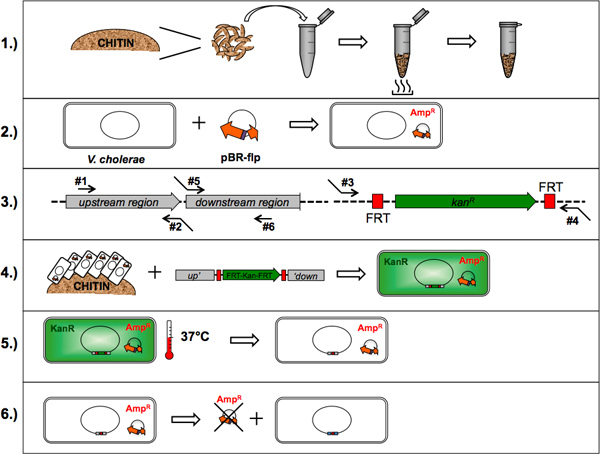
Figure 1. Flow chart of the TransFLP method. The six points described in this protocol are drafted: 1.) Preparation of chitin flakes; 2.) Optional: Artificial transformation of V. cholerae with Flp recombinase-encoding plasmid; 3.) Oligonucleotide design and polymerase chain reaction; 4.) Chitin-induced natural transformation; 5.) Removal of selective cassette(s) by flp-recombination; 6.) Plasmid curing. Click here to view larger figure.
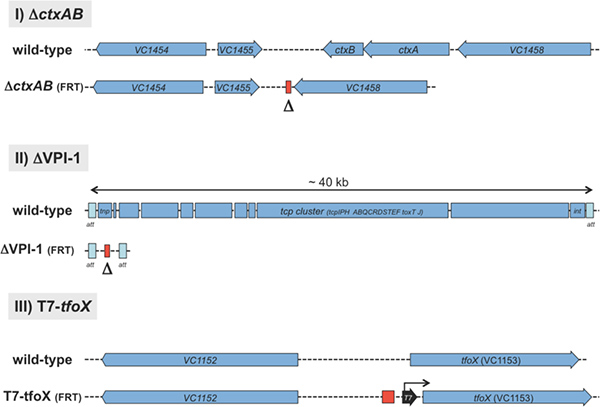
Figure 2. Schematic representation of three application possibilities for the TransFLP method. I) A 1458 bp DNA fragment, containing the cholera toxin encoding genes ctxA and ctxB, was deleted as indicated by Δ. II) The ~ 40 kb Vibrio pathogenicity island 1 (VPI-113) was deleted from the wild-type strain. III) A T7 RNA polymerase-dependent promoter sequence (28 bp) was integrated upstream of the tfoX gene. The red rectangle indicates the left behind short nucleotide sequence of a single FRT site10. Click here to view larger figure.
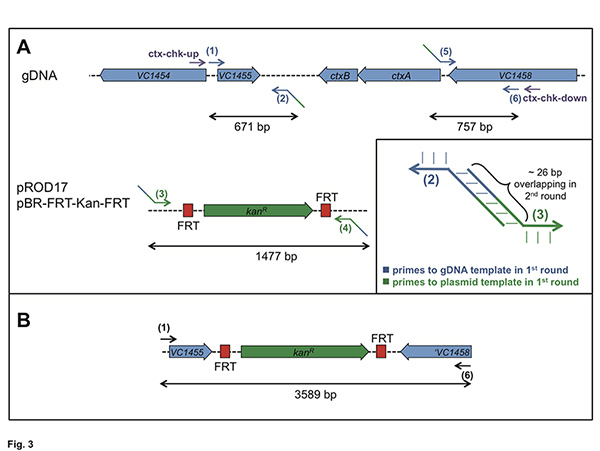
Figure 3. Explanation for primer design and performance of PCR based on the deletion of ctxAB. Panel A: For the 1st round of PCR genomic DNA (gDNA) and plasmids pROD1712 and pBR-FRT-Kan-FRT, respectively, served as templates. The required oligonucleotides are #1 to #6. The primer #2 / #3 and #4 / #5 (example in inset) should also possess significant overlap to each other at their 5’ends. Panel B: After the 2nd round of PCR, which is based on the 1st round PCR fragments as templates, a long fragment should be obtained using primer pair #1 + #6. Click here to view larger figure.
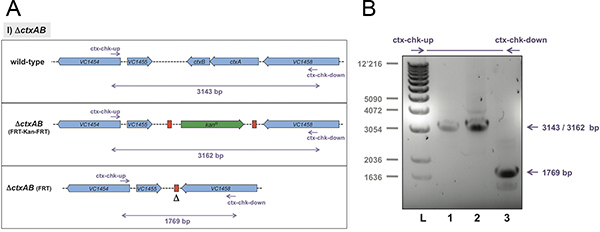
Figure 4. Expected result exemplified for the ctxAB deletion strategy. The original strain (wild-type), the insertional mutant ΔctxAB::FRT-Kan-FRT, and the final strain ΔctxAB::FRT were tested for their genotypes. Panel A: Schematic representation of the differentiation power of the primer pair ctx-chk-up and ctx-chk-down, most notably with respect to the excision of the antibiotic cassette. Panel B: Whole bacterial cells were used as template in PCR reactions. To check for the changed DNA locus primers chk-up and chk-down were utilized. The amplified PCR fragments were separated by agarose gel electrophoresis and visualized using SYBR safe DNA stain. Lane 1: Wild-type V. cholerae strain; lane 2: strain ΔctxAB::FRT-Kan-FRT; lane 3: ΔctxAB::FRT. Expected PCR fragment sizes according to panel A are indicated on the right. L, 1kb ladder (Invitrogen). Click here to view larger figure.
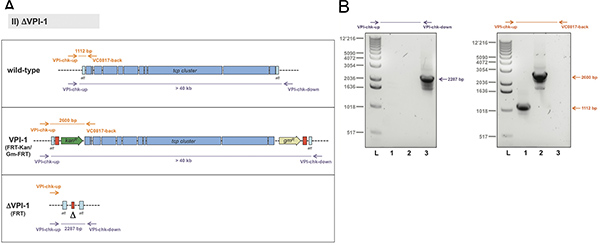
Figure 5. Strategy to verify the deletion of the genomic island. Panel A: Scheme of Vibrio pathogenicity island 1 (VPI-1) (freely adapted from Ref.16; not to scale). For the wild-type strain the region of interest is non-amplifiable by means of standard PCR and the VPI-1-specific primers chk-up and chk-down (~ 40 kb). Therefore an additional oligonucleotide annealing within the region to-be-deleted was applied (VC0817-back) together with VPI-chk-up. Panel B: Representative result using genomic DNA of the respective strains as template and the primer pairs indicated above each image. Strains tested: Wild-type V. cholerae strain (lane 1), strain VPI::FRT-Kan/Gm-FRT (lane 2), and ΔVPI-1::FRT (lane 3). Sizes of the PCR fragments as predicted in panel A are indicated on the right. L, 1kb ladder (Invitrogen). Click here to view larger figure.
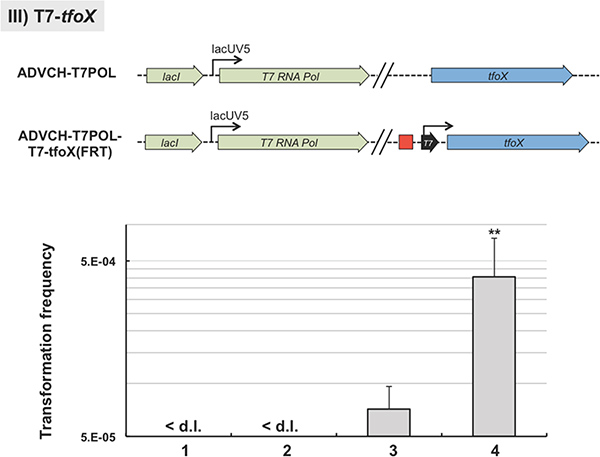
Figure 6. Representative phenotype after insertion of an artificial promoter sequence. The T7 RNA polymerase-dependent promoter sequence was integrated upstream of the competence regulatory gene tfoX using the TransFLP method (sketch above graph). The parental V. cholerae strain (ADVCH-T7POL) contains a T7 RNA polymerase gene driven by the lacUV5 promoter. Graph: The parental strain ADVCH-T7POL (lanes 1 and 2) and the strain manipulated by TransFLP (ADVCH-T7POL-T7-tfoX::FRT; lanes 3 and 4) were tested for natural transformability in the absence (lanes 1 and 3) or presence of 1mM IPTG (lanes 2 and 4). Transforming genomic DNA was derived from strain A1552-LacZ-Kan8. Transformation frequencies are given on the Y-axis. < d.l., below detection limit of ~5 x 10-8. **P < 0.01 (as determined by student’s t test of log-transformed data). Black arrow with text ‘T7’: T7 RNA polymerase-dependent promoter sequence. Click here to view larger figure.
