Part 1: Preparation of gold-coated capillaries for nanoflow electrospray ionization
Analysis of non-covalent complexes is usually performed by means of nanoflow electrospray ionization (nESI)6, using glass or quartz capillaries which have been pulled to a fine tip (~1 μm inner diameter), and coated with conductive material (usually gold). Such capillaries are available ready-to-use from commercial sources (New Objective or Proxeon); however, it can be more cost-effective to prepare them in-house:
- Stick two strips of a double-sided adhesive pad to the bottom of a Petri dish, 2 cm apart. Place a glass rod (8 cm x 5 mm) in the center of one of the pads. The adhesive pad will hold the capillaries in place, and the glass rod will support the prepared capillaries, and keep the tips from breaking (Figure 1).
- Use borosilicate glass capillaries, 1.0 mm OD x 0.78 mm i.d. (we use packs of 500 thin wall borosilicate capillaries from Warner Instruments, cat. no. G100TF- 4). Insert one capillary into the needle puller (we use model P-97 from the Sutter Instrument Co.). Clamp the capillary gently in place, and adjust its position so that it lies in the center of the needle puller's heating filament. Tighten the clamps gently, until the capillary is held firm at both ends.
- Pull the capillary, using a predefined program. Every capillary pulled will give rise to two final, shaped capillaries. The process of programming the puller is one of trial-and-error, until an acceptable tip shape is obtained. We use the following program:
Step Heat Pull Vell Time 1 750 – 15 80 2 700 – 15 50 3 750 200 20 80 - Remove the pulled capillaries from the instrument and inspect the tips, discarding any that are deformed or broken. Use blunt tweezers (we use positioning tweezers from C.K. Precision), to place the capillaries in the Petri dish. The base of the capillary should be attached to the adhesive pad and the upper part should lean on the glass rod, with the tip pointing up.
- Once the Petri dish is full (about 80 capillaries fit into a 10 cm-diameter dish), insert the plate into the gold sputter coater (we use model no. EMS550, from EMS). Make sure that the gas supply is chosen according to the manufacturer's instructions, and activate a predefined coating cycle (we use Argon pressure 4 psi, at a vacuum pressure of 5 x 10-2 mbar, a current of 45mA, and a coating time of 1 min, for 3-6 cycles, until the capillaries are evenly golden).
Part 2: Sample preparation
- Low micromolar concentrations of sample are required (1 – 20 μM). If necessary, concentrate the sample using centrifugal ultrafiltration devices (e.g., Vivaspin from Sartorius, or NanoSep from Pall Corporation). It is recommended that you verify the adsorption of the protein complex to the device membrane, before use.
- Often, the purification buffers or storage solutions of the protein complex are not compatible with nESI, for which only volatile solutions may be used. Therefore, buffer exchange is necessary. This critical step, in which all traces of salts, buffer molecules or any other non-volatile adducts such as glycerol, DTT, or EDTA are removed, determines the quality of the spectra. Usually, aqueous ammonium acetate solution is used, at a concentration of between 5 mM and 1 M, and at 6-8 pH. Buffer exchange can be performed using a microcentrifuge gel filtration column (e.g., Micro Bio-Spin 6 chromatography columns from Bio-Rad). This step can be repeated 1-3 times with minimal dilution (less than a factor of 1.3 per device5), until maximal exchange is achieved. If both concentration and buffer exchange are required, these may be done together, using centrifugal ultrafiltration (e,g., Vivaspin from Sartorius, or NanoSep from Pall Corporation).
Part 3: Calibrating the mass spectrometer for high mass measurements
Most of the experiments conducted on multi-protein complexes are performed using a nano electrospray quadrupole-time-of-flight (Q-ToF) instrument. It is suggested that you use a quadrupole mass filter adjusted to low frequencies, to enable transmission and mass analysis of ions with high m/z values7,8. It is also recommended that gas inlets7,8 or sleeves9 be added to the instrument in the first ion guide, to enable pressure control at the first vacuum stage. The latter enables optimization of the transmission, and desolvation of very large ions7-9. Currently, commercial ESI-ToF and Q-ToF instruments are available from several manufacturers (for example, Waters, SCIEX, Bruker, or Agilent) which can be modified relatively easily and cost-effectively, for native MS applications7,8. It is possible, however, to use standard ToF or QToF configurations on instruments such as LCT or QToF1 (Waters) to acquire mass spectra for complexes up to 1 MDa, without the need for hardware modifications5.
The protocol outlined below was conducted on a Synapt instrument (Waters).
- Prepare 100 mg/ml solution of CsI in purified water. CsI is used for high mass calibration as the singly charged salt clusters, (CsI)nCs+ extend over a wide mass range, from 393 m/z to well over 10,000 (Figure 2).
- Using blunt tweezers, take a coated capillary from the Petri dish and load 2μL of the CsI solution into the capillary, using an Eppendorf GeLoader tip.
- Insert the capillary into the capillary holder, and adjust the capillary in such a way that the tip is approximately 10 mm away from the edge of the holder.
- Slide the solution towards the tip of the capillary either manually, or using a spin down adaptor.
- Place the capillary under an optical microscope and trim the tip, using the sharp AA-type tweezers.
- Connect the capillary holder to the nanoflow ES interface. Rotate the x-y-z stage backwards to avoid damaging the capillary, and push the stage into its activated position. The capillary should be placed 1 – 10 mm from the cone orifice.
- Apply capillary voltage (1,050-1,400 V) and low nanoflow pressure (0.00-0.03 bar) until spray is initiated, then try to reduce the nanoflow pressure to a minimal value.
- Optimize the signal intensity by adjusting the location of the x-y-z stage, the capillary voltage, the nanoflow pressure, and the desolvation gas flow.
- To detect a wide mass range of CsI peak series, the accelerating voltages should be optimized (we use the following parameters: capillary 1.3-1.7 kV, sample cone 80-150 V, extraction cone 1-3 V).
- To optimize the transmission of high mass ions, which require gentle desolvation conditions, the backing pressure in the initial vacuum stage, between the source and the analyzer, should be raised. This can be achieved by carefully reducing the conductance of the source vacuum line to the scroll pump, by partially closing the isolation valve (SpeediValve). In order to define the optimal point, the latter should be done while monitoring the effect on signal intensity (we usually use 3.0-6.5 mBar).
- Collect about 30 scans at 1 scan/sec, at an m/z range of between 1,000 to 15,000 m/z.
- After acquisition, calibrate the TOF using the appropriate calibration table.
Part 4: MS analysis of intact protein complexes
- Load your sample, as described (Part 3, Sections 2-5), and initiate spray.
- Start by initial optimization of the spray (Part 3, Sections 6-9), until the signal is detected. The exact position of the charge states is protein-dependent; however, it can be predicted, using the relationship between ion mass and average charge state, thus: (Zav)10: Zav = 0.0778√(m) , in which m is the mass of the complex in Daltons. To prevent complex dissociation, do not heat the ion source: either switch the heater off, or keep the temperature below 40 °C.
- Vary the capillary position, sample cone and extractor voltages to maximize ion transmission, and check the resulting change in the spectra. A possible starting point could be cone voltage 100 V; extractor cone: 1 V; capillary voltage 1.5 kV. Optimize these parameters, in combination with the nanoflow pressure.
- To improve desolvation and strip off residual water and buffer components, increase the bias voltage and the gas pressure in the collision cell. This step should be performed carefully, to prevent dissociation of the complex. Typical bias voltages are within the range of 10-100 V, with a trap gas flow of 1-10 ml/min (Figure 3). Manual adjustment of the RF setting and quadrupole profile may improve the transmission of high mass ions.
- Adjust Trap and Transfer collision energies. Often, higher voltages are required for transmission of high mass ions (usually within the range of 10-30 V). At this point, it is important to avoid collision-induced dissociation of the complex.
- After a stable signal is reached, it is recommended that the nanoflow pressure and capillary voltage be decreased to minimal values, while maintaining stable spray.
Part 5: Tandem mass spectrometry: dissociating protein complexes
- Once an optimum and stable signal has been obtained, select a precursor ion. Set the mass center and isolation width (we generally use a LM resolution of 12, and a HM resolution of between 13 and 15). Use a wide mass range to detect the high mass/low charge dissociation products. It is recommended that you set the m/z range to the maximum level, and then reduce it to the desired values. We further suggest superimposing the MS and MS/MS spectra, to validate the isolation of the chosen precursor ion.
- To perform MS/MS, dissociate the precursor ion by increasing the collision energy (CE) and the pressure on the collision cell. Increase either Trap or Transfer CE gradually, in steps of 10-20 V, and elevate the collision gas pressure to 0-5 ml/min (common values). Monitor changes in the spectra until optimal activation conditions are reached. High activation energy may induce the dissociation of one or more subunits from the intact complex, and clarify the interaction affinities of different subunits. We typically use Argon as a collision gas in the Trap/Transfer cells, although benefits of using a heavier gas (e.g., Xe or SF6) have been reported; however, these gases are substantially more expensive11 (Figure 3).
- It is recommended that more than one charge state be selected for MS/MS analysis. In the case of overlapping components, acquiring a set of tandem MS spectra will assist in resolving the charge series of the different populations. Moreover, higher charge states will dissociate more easily, compared to lower charge states12.
- In addition to the characterization of the intact complex, it is suggested that smaller subcomplexes in solution will be generated, under mild denaturing conditions. The MS and MS/MS analyses of subcomplexes form the basis for defining the subunit architecture of the complex13. For partial disruption of subunit-subunit interactions, gradually add organic solvents (e.g. methanol, isopropanol, or acetonitrile) up to a concentration of 50%, or change the pH of the solution by adding ammonia or formic acid (up to a concentration of 4%).
- To determine the masses of the individual subunits that compose the complex, it is important to acquire a spectrum under denaturing conditions. This can be carried out using Zip-Tip C4 (Millipore) with a 25:75 water/acetonitrile ratio, with 1% formic acid as the elution solvent.
Part 6: Data processing and analysis
- Data is analyzed offline, using a spectral analysis programs for MS spectra. We normally use the MassLynx program (Waters).
- For spectra that span a wide m/z range, we recommend that different schemes of smoothing and centroid parameters be applied for the high and low m/z regions, to reflect the difference in peak resolution of these regions.
- To identify charge series and calculate charge states and masses, the "manual find" function of MassLynx can be applied. In cases of complex mass spectra with overlapping components, manual calculation of masses may be easier.
- To facilitate these assignment processes, additional software may be used; for example: MaxEnt for spectra deconvolution14, SOMMS for peak fitting and simulation15, and SUMMIT for assigning the composition and stoichiometry of protein subcomplexes and generating protein interaction networks13,16,17.
Part 7: Representative Results

Figure 1. Preparing gold-coated nano-electrospray capillaries.
A. Attach two double-sided adhesive strips to a Petri dish, 2 cm apart. To support the prepared capillaries, place a glass rod (8 cm x 5 mm) in the center of one of the pads. B. Stick the blunt end of the prepared capillaries to the adhesive pad, and lean the tip on the glass rod. C. Once the Petri dish is filled with the prepared capillaries, coat them with gold until a thin film of gold is evenly deposited on the external surface of the capillaries.
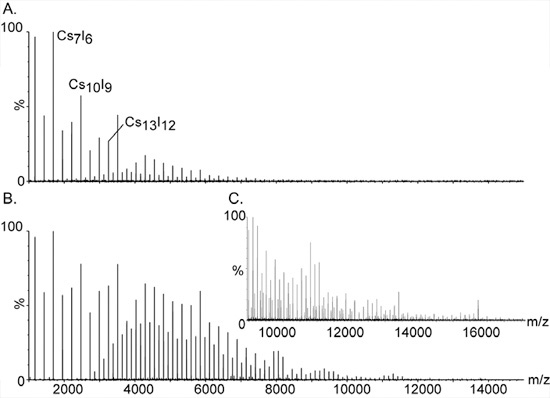
Figure 2. High mass calibration using cesium iodide ions.
The large and monisotopic clusters of CsI have made it the compound of choice for calibrating mass spectrometers for high mass analysis. The series of equally spaced peaks extend over a wide range, from m/z 393 to well over 10,000. They are assigned to singly charged salt clusters of the general composition (CsI)nCs+. Additional signals between the major peaks are caused by double- and triple-charged species of the series; [(CsI)nCs2]2+ and [(CsI)nCs3]3+, respectively. Increasing the pressure at the initial vacuum stage is essential for detecting the high mass clusters. The effect of pressure on the high mass peaks is demonstrated in Panels A. and B. with pressure readbacks of 1.2 and 5.3, respectively. C. Expansion of the mass spectrum shown at B.
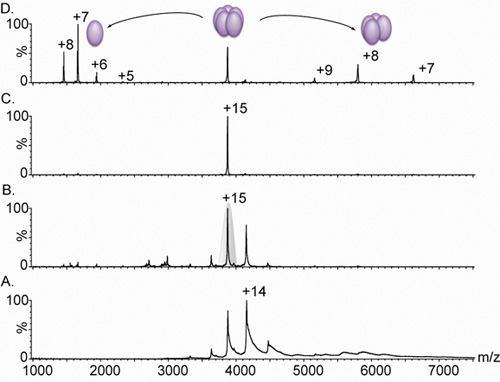
Figure 3. Nanoflow electrospray mass spectra of a pentameric lectin.
A. Mass spectrometry of a lectin variant complex (derived from Lib1-B7 by directed evolution18) gives rise to a charge state distributions between 3,000 and 5,000 m/z; however, due to inadequate desolvation of the ions, the peaks are broad. Comparison of panel A. and B. show the effect of increasing the bias voltage from 4 V (A.) to 15 V (B.) on the peak width. This increase in accelerating conditions causes the stripping of residual water and buffer components, yielding a highly resolved spectrum. The measured mass (60,240 ± 38 Da) corresponds to a pentameric complex. C. The +15 charge state was then selected for tandem MS analysis (shaded in grey in Panel B.) D. The increase in collision energy causes the release of a highly charged monomer, centered at 1,664 m/z, and a stripped tetrameric complex, in the range of 5,000–8,000 m/z. All spectra were obtained from a sample containing 20 μM of solution in 0.5 M ammonium acetate.
