MiRNAs regulate cellular processes by binding to target mRNAs. Therefore, identifying mRNA targets are a key to understand miRNA function. Here we elaborate a method for identifying mRNA target of cellular miRNA. This protocol is adapted from Wani et al.35 with the modification of utilizing biotinylated LNA-based miRNA mimics to pulldown target mRNA. The use of LNA-based oligonucleotide mimics enhances specificity of target binding owing to the higher stability of the modified ribose ring without toxic effects36,37,38. We employed this method to pull down PARP-1 mRNA as the target of cellular miRNA miR-125b. The highest level of miR-125b expression is detected in brain tissue that regulates neuronal function39,40. In an attempt to identify the targets of miR-125b in neuronal cells, recently we reported that miR-125b negatively regulates PARP-1 expression by binding to the 3' UTR of PARP-1 mRNA41.
Pulldown of miRNA:mRNA Complex
We used HEK-293T cells to study the interaction between miR-125b and PARP-1 mRNA, since these cells are widely used in cellular, biochemical, and molecular biology studies. A schematic of the protocol used for the mRNA target is depicted in Figure 1. First, HEK-293T cells (4 x 105–5 x 105 cells per well) were seeded overnight in a 6-well tissue culture plate and incubated at 37 °C with 5% CO2 for 18–24 h. Thereafter, 75 picomoles of 3'-biotinylated miR-125b mimics and 3'-biotinylated scrambled controls were transfected into the cells using liposome based transfection method. Transfected cells were incubated at 37 °C and 5% CO2 for an additional 36 h and harvested. Thereafter cellular lysates of transfected cells were prepared by the freeze-thaw lysis method. The freshly prepared cellular lysates were incubated with the blocked streptavidin coated magnetic beads on a bench top nutating mixer for 1 h at room temperature. Following this, the beads were processed and washed using freshly prepared ice cold pull-down wash buffer on the magnetic separator. Finally, the beads were resuspended in 100 µL of nuclease free water for further analysis.
Detection of Target mRNA in the pull-down miRNA:mRNA Complex
To detect the target mRNA of miR-125b from the pull-down mixture, we employed a qPCR assay (Figure 1). We designed forward and reverse primers (FP and RP) within the ORF to amplify PARP-1 mRNA (Figure 2, Table 5b). We also designed primers sets for detecting p53 mRNA, a known target of miR-125b42, as a positive control and included primers to amplify actin mRNA as a non-specific negative control. In addition, to further confirm the specificity of amplification, we designed primers for detecting the 3' UTR regions of PARP-1, p53 and actin. We performed qPCR using the methods described in the protocol (section 6).
Figure 3 shows the qPCR based amplification of target mRNA's relative to scrambled controls from the purified beads. The level of PARP-1 mRNA was markedly higher in samples pulled-down with biotinylated miR-125b mimics when compared to the scrambled controls. As expected, higher mRNA levels of the positive control p53 mRNA and minimal amplification of the negative control actin mRNA was observed in these samples. Similar levels of amplification were obtained using the primers targeting the 3' UTR of PARP-1 and p53 compared to the negative control actin (Figure 3B). The qPCR results of the ORF regions (Figure 3A) and the 3'UTR regions (Figure 3B) of PARP-1 mRNA and p53 mRNA showed some levels of variability. Several factors including binding affinity, number of miRNA binding sites, and differences in primer sequence dependent amplification may contribute to the variable levels of amplification in Figure 3. Nevertheless, these data strongly support the feasibility and specificity of the method for validating mRNA targets of cellular miRNAs.
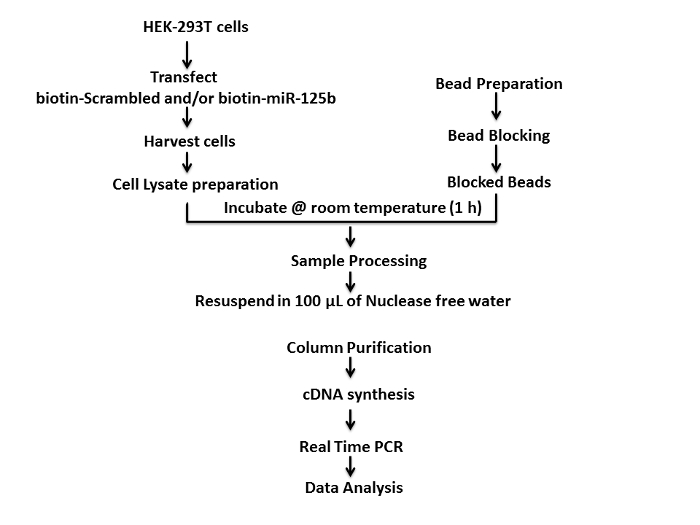
Figure 1: Schematic representation of the protocol for the target capture assay using biotinylated-miRNA mimics. Please click here to view a larger version of this figure.
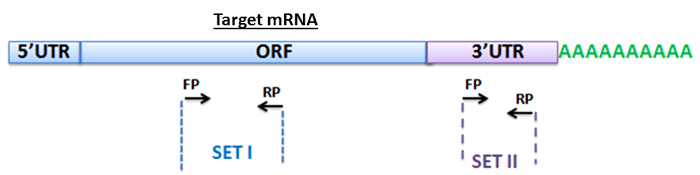
Figure 2: Primer design for amplification of mRNA targets. Schematic representation of the primers used to detect the target mRNA of miR-125b. One set of primers (Set I) was designed within the ORF region of the transcripts and the other set of primers (Set II) was designed in the 3' UTR region of the target genes. FP and RP represent forward and reverse primers for each set. Please click here to view a larger version of this figure.
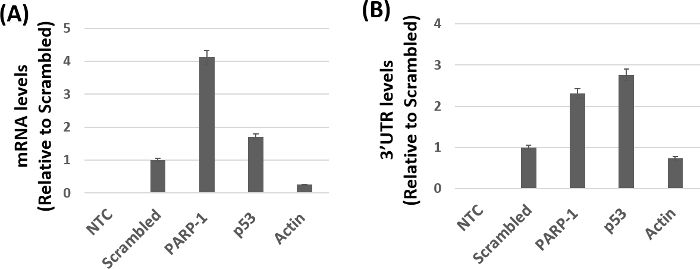
Figure 3: miR-125b target identification by qPCR. (A) mRNA levels using primer set I, which amplifies the ORF region of target mRNA. Higher expression levels of PARP-1 and p53 mRNA (positive control) were observed when compared to actin mRNA (negative control). (B) Amplification of the 3' UTR region of target mRNA using primer set II. No amplification was observed in the no template controls (NTC). All reactions were performed in triplicates and the data was analyzed using the appropriate data analysis software based on the 2-ΔCt method. Error bars represent standard error of the mean. Please click here to view a larger version of this figure.
| Component | Stock | Final volume for 20 μL reaction |
| Template (RNA) | 50 ng/µL | 1.0 μL |
| Reaction buffer | 5x | 4.0 μL |
| Oligo dT primer | Master stock | 2 μL |
| Reverse Transcriptase | Master stock | 1 μL |
| Nuclease-free distilled water | – | 12 μL |
| Total Reaction Volume | 20 μL |
Table 1: cDNA synthesis reaction mixture.
| 105 °C = 30 s |
| 42 °C = 60 min |
| 95 °C = 5 min |
| 10 °C = Hold |
Table 2: cDNA synthesis thermal cycling conditions.
| Component | Stock | Final volume for 10 μL reaction |
| cDNA product | – | 1.0 μL |
| iTaq Universal SYBR mix | 2x | 5.0 μL |
| Target (ORF/3’ UTR) F.P. | 10 μM | 0.5 μL |
| Target (ORF/3’ UTR) R.P. | 10 μM | 0.5 μL |
| Nuclease-free distilled water | – | 3 μL |
| Total Reaction Volume | 10 μL |
Table 3: qPCR reaction mixture.
| 95 °C = 10 min |
| 30 cycles of: |
| 95 °C = 10 s |
| 56 °C = 30 s |
| 72 °C = 30 s |
| Melt Curve Analysis |
| 65 °C= 31 s |
| Linear Ramp rate = 0.5 °C/s |
| Acquisition = 0.5 °C intervals |
Table 4: qPCR thermal cycling conditions.
| (A): Biotinylated Oligos | ||
| Biotinlyated Oligos | Stock | Sequence (5' to 3’) |
| hsa-miR-125b | 25 μM | UCCCUGAGACCCUAACUUGUGA-Biotin |
| Scrambled Control | 25 μM | GAUGGCAUUCGAUCAGUUCUA-Biotin |
| (B) Primers | ||
| Target Primer (ORF) | Stock | Sequence (5’ to 3’) |
| PARP-1 (FP) | 100 μM | GAGGTGGATGGGTTCTCTGA |
| PARP-1 (RP) | 100 μM | ACACCCCTTGCACGTACTTC |
| p53 (FP) | 100 μM | TGTGACTTGCACGTACTCCC |
| p53 (RP) | 100 μM | ACCATCGCTATCTGAGCAGC |
| ACTIN (FP) | 100 μM | GCTCGTCGTCGACAACGGCTC |
| ACTIN (RP) | 100 μM | CAAACATGATCTGGGTCATCTT |
| (C) Primers | ||
| Target Primer (3’ UTR) | Stock | Sequence (5’ to 3’) |
| PARP-1 (FP) | 100 μM | ATTGGGAGAGGTAGCCGAGT |
| PARP-1 (RP) | 100 μM | CTACCCATCAGCAACTTAGCG |
| p53 (FP) | 100 μM | GGCCCATATCTGTGAAATGC |
| p53 (RP) | 100 μM | CTGCAGGAAGGCAGGTTTT |
Table 5: Oligonucleotide Names and Sequences.
