To illustrate the capabilities of the openFLIM-HCA instrumentation, we present three exemplar FRET applications. The first concerns FRET constructs that are adapted from FRET model constructs developed by Stephen Vogel's laboratory38. They comprise a series of genetically expressible fluorescent constructs in which a donor fluorescent protein (mTurquoise) is linked to an acceptor fluorophore (Venus) by short controlled lengths of 5, 17 and 32 amino acids (mTq5V, mTq17V, mTq32V). The different linker lengths between donor and acceptor fluorophores result in different FRET efficiencies and therefore different donor lifetimes. To provide a negative control, the Venus in the short 5-amino acid linker construct is replaced by Amber — a non-fluorescent mutation of YFP (mTq5A) that cannot act as a FRET acceptor38. We replaced Cerulean in the original pCXV and pC5A vectors by restriction digesting the vectors with NheI and BglII enzymes and ligating mTurquoise fragments digested using the same enzymes from the pmTurquoise-N1 vector. The linker region was unmodified by this substitution.
Figure 6 presents an exemplar FLIM FRET assay using this model system expressed in Cos cells, in which a multiwell plate is arrayed with 3 columns each of cells transfected with the negative control (columns 1-3), the 5-amino acid linker FRET construct (columns 4-6), the 17-amino acid linker FRET construct (columns 7-9) and the 32 amino acid linker FRET construct (columns 10-12). Row H contains wells for TVB estimation. Figure 6(a) shows examples of automatically acquired fluorescence lifetime images of typical fields of view in each well. It is apparent that the negative control (columns 1-3) present the longest lifetimes and that the donor lifetime is lowest for the FRET construct with the shortest linker length (columns 4-6). Figure 6(b) shows how the mean lifetime averaged over all the FOVs in each well varies across the multiwell plate. Figures 6(c) and (d) show exemplar donor fluorescence intensity-merged lifetime maps for a FOV of mTq5A and mTq17V respectively. Figure 6(e) shows the donor fluorescence intensity decay profile averaged over all the cells imaged in well A1, together with the fit to a monoexponential decay model and the IRF (which is dominated by the GOI gate width). Figure 6(f) presents the average fluorescence lifetime for each column of the multiwell plate obtained from a pixel-wise monoexponential fit. A table of the mean lifetime and standard deviation (STD) can be found in the Table 1 below. The data acquisition for the images shown in Figure 6 took approximately 160 min to acquire. The analysis took 92 s on a 2.6 GHz computer with 10 cores and 64 GB of RAM.
The second exemplar assay demonstrates the application of FLIM FRET to read out the oligomerization of HIV-1 Gag during the assembly of HIV-1 virions as a means to test inhibitors of this process25,42. Expressing HIV-1 Gag in appropriate host cells provides a model system for this stage of the HIV-1 lifecycle since it leads to the formation of viral-like particles (VLPs) that are similar to immature HIV-1 virions. For this assay, we expressed HIV-1 Gag protein fused with CFP in HeLa cells and compared the action of different inhibitors via their impact on the FRET signal that resulted from the Gag oligomerization. Details of the inhibitors used are provided in reference 42.
The details of the sample preparation and experimental measurements are described comprehensively in reference 25, where we demonstrated that we could use FLIM to read out both heteroFRET between aggregating Gag proteins labeled stochastically with CFP and with YFP, and also homoFRET in cells expressing only Gag labeled with CFP. Although homoFRET does not usually present a change in donor lifetime, it does for the special case of CFP where the fluorophore exists in two isomers with different mean fluorescence lifetimes40,41. The CFP homo-FRET readout has the advantages of simplified sample preparation compared to CFP/YFP hetero-FRET and is more spectral efficient, which could be important for multiplexed FRET readouts.
Our previous work applied FLIM FRET to assay the Gag oligomerization in the presence of an N-myristoyltransferase (NMT) inhibitor42. This inhibitor disrupts the endogenous enzymes responsible for the addition of myristic acid to Gag, which enables Gag proteins to bind to the plasma membrane and is a prerequisite43 in the formation of HIV virions or VLPs. We showed that both heteroFRET and homoFRET readouts could achieve Z' of >0.6 in dose response studies with an NMT inhibitor. Z' is a parameter used in the pharmaceutical industry to indicate the quality of an assay and represents the differences in the mean values of the positive and negative controls and their relative spreads to generate a single value metric of assay quality — with Z' > 0.5 being considered "excellent" for a pharmaceutical assay44. We note that this excellent performance was achieved with samples for which the change in mean donor lifetime was of the order of 300 ps — demonstrating the statistical power of analyzing FLIM data for large numbers of cells, which enables useful readouts to be realized using much smaller changes in fluorescence lifetime than is possible with the typical numbers of cells imaged in manual microscopy experiments.
Here, we present a multiwell plate FLIM FRET data set comparing 4 inhibitor compounds for HIV Gag oligomerization (designated ICL13, ICL14, ICL15 & ICL16). For this experiment, we utilized the homoFRET readout with HeLa cells transiently transfected with the Gag-CFP plasmid. A total of nine different doses of each inhibitor were applied following the standard protocol outlined in reference 32, allowing two repeat wells per condition. As a positive control, column 1 presents cells expressing myr-Gag, a mutated Gag protein lacking the myristic acid moiety that cannot form VLPs and so simulates total inhibition. A negative control was provided by dosing cells expressing Gag-CFP only with just the vehicle used to deliver the inhibitors in the other columns (column 11). A total of eight FOV were acquired per well.
Data was fitted with a single exponential decay model on a pixel-by-pixel basis and the fluorescence lifetime plate map showing the average lifetime per well (over all pixels above the intensity threshold across the eight fields of view) is shown below in Figure 7(a). Figure 7(b) shows the corresponding plot of fluorescence lifetime as a function of inhibitor concentration. Three of the inhibitors show an inhibitory effect (ICL13, ICL15 & ICL16) when incubated with cells expressing Gag-CFP. One inhibitor (ICL14) shows no effect at any dose tested. The Z' calculated for this plate was 0.51. The values obtained for the positive and negative controls are shown on Figure 7(b) as the points in black at 100 µM and 0.1 nM.
The dose response curves for ICL13, ICL15 and ICL16 shown in Figure 7(b) were then fitted to the 4 parameter logistic nonlinear regression model with the Hill coefficient fixed to 145 with the maximum and minimum lifetimes fixed to the values obtained from the positive (column 1) and negative (column 11) controls respectively. The returned IC50 values for the three curves were then: 38 nM, 24 nM and 116 nM for ICL13, ICL15 and ICL16 respectively. For comparison with IC50 values obtained through intracellular FLIM FRET measurements, we determined IC50 for inhibition of enzymatic activity of recombinant human NMT1 in our previously reported biochemical enzyme assay42. The three compounds found to be active by FLIM FRET are also the most active in this assay (IC50 values of 17 nM, 51 nM, and 39 nM for ICL13, ICL15 and ICL16, respectively), with ICL14, which showed no significant response in the FLIM FRET assay, being ca. 100-fold less active against NMT1 (IC50 of 1700 nM). For the active compounds, the IC50 values obtained through these independent assay modalities are remarkably similar, with minor variations likely attributable to differences in uptake or metabolism of the compound in cells.
This small study highlights the ability of FLIM HCA to discriminate the potency of different compounds which act on the same target of interest. We believe it is the first demonstration of a screen using automated FLIM in a high content context to generate multiple dose response curves and illustrates the potential for FLIM to be applied to screen for and/or characterize useful therapeutic compounds.
The third exemplar FLIM FRET experiment concerns a genetically expressed FRET biosensor for Exchange protein activated by cyclic adenosine monophosphate (cAMP (EPAC)), which is a Rap-1 guanine exchange factor that binds specifically to cAMP. This EPAC FRET biosensor (EPAC-SH188) utilizes mTurquoise2 fluorescent protein (mTq2FP) as the donor fluorophore and incorporates two Venus-FP to implement a composite acceptor46. cAMP is a ubiquitous second messenger and is involved in a plethora of intracellular processes throughout many different organisms. It is synthesized by adenylate cyclase from the conversion of ATP at the cell membrane. Here we demonstrate our ability to read out and quantify its activity in live HEK293T cells following the addition of forskolin, an adenylate cyclase activator that upregulates intracellular production of cAMP and therefore increases its cytoplasmic concentration. This EPAC sensor undergoes FRET in its inactivated (unbound) state and its donor lifetime increases with cAMP concentration as the cAMP leads to the opening of the biosensor. The mono-exponential decay profile of mTq2FP makes this biosensor better suited for quantitative lifetime analysis than CFP-based biosensors 45. Figure 8 shows an example of a time course recorded using the automated multiwell plate FLIM microscope where we have plotted the change in mean lifetime and the change in the FRETing donor population fraction over time following the addition of 100 µM forskolin. For simplicity, we assume that the total signal can be described by a mixture of monoexponential FRETing and non-FRETing donors and the population fraction of the activated EPAC FRET biosensor was obtained by fitting a double exponential decay profile across the time series.
For the data acquisition of cAMP (EPAC) five gate delays, with a gate width of 4,000 ps, were chosen with the delay time set to 1,300 ps, 4,300 ps (peak intensity), 4,919 ps, 6,656 ps, 8,035 ps, 1,0391 ps and 16,000 ps. The camera integration time for each delay was 250 ms. At one well, we acquired a FLIM time-course with images acquired every 10 s over 10 min. The data points acquired at times 120 s and 130 s were removed because the addition of forskolin caused a small shift in the focal position of the sample and therefore of the fluorescence intensity recorded during the FLIM acquisitions at these time points.
For the data analysis the images were loaded into FLIMfit and segmented per cell. The fluorescence lifetime data was fitted to a double exponential decay model using an IRF and a fixed t0 value obtained from a reference dye (75 µM Coumarin 6). The donor fluorescence mean lifetime averaged over all cells is shown in Figure 8(a). Figure 8(b) shows the change in FRETing population fraction over time calculated by fitting the same FLIM data to the same double-exponential decay model
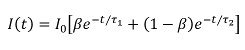
where β is the FRETing donor fraction. We assume a mixture of FRETing and non-FRETing elements for the time points prior to addition of forskolin. To obtain these lifetimes, a double exponential fit was applied to the first three time points giving lifetimes of τ1 = 3,964 ± 21 ps for the non-FRETing donor, and τ2 = 3,022 ± 27 ps for the FRETing donor. These were fixed for the subsequent fit of the whole data set to obtain the β values at each time point.
In summary, we have presented exemplar FLIM HCA results for three experimental samples. The first was a 96 well plate arrayed with Cos cells expressing FRET constructs comprising a mTqFP donor linked to either a Venus FP acceptor or a non-fluorescent Amber construct by varying lengths of peptide linker. The change in FRET efficiency and therefore donor lifetime with linker length is clearly apparent and the standard deviation of the mean lifetime for each construct was less than 36 ps. The second exemplar assay was a "screen" of four potential inhibitors for myristoylation of the HIV Gag protein for which the % inhibition was calculated from the variation of mean donor fluorescence lifetime with inhibitor concentration measured in Hela cells expressing Gag protein labeled with CFP. This readout is based on homoFRET, which would not usually present a change in donor lifetime but does for CFP because this exists in multiple isomers with different fluorescent lifetimes. This can be useful in terms of spectral efficiency, e.g., for multiplexing different FRET readouts25. The third exemplar assay was a time course monitoring live HEK293T cells expressing the cAMP FRET biosensor, EPAC. This showed the expected response following treatment with forskolin and the application of FLIMfit using a double exponential decay model with the number of detected photons being commensurate with live cell imaging — as we have previously shown with the LIBRA FRET biosensor for IP347.
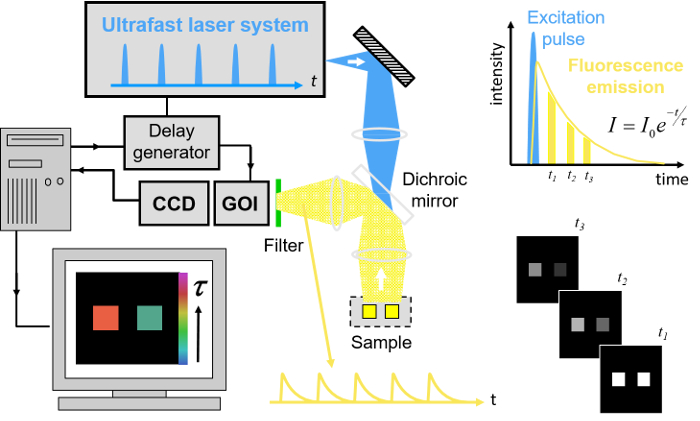
Figure 1. Schematic of wide-field time-gated FLIM using a gated optical image intensifier (GOI). Left hand side, shows a schematic of the experimental system. Top right, schematic of excitation pulse, position of time-gated images and monoexponential fit to data. Bottom right, cartoon showing the fluorescence decay from the two objects shown in the experimental system. Please click here to view a larger version of this figure.
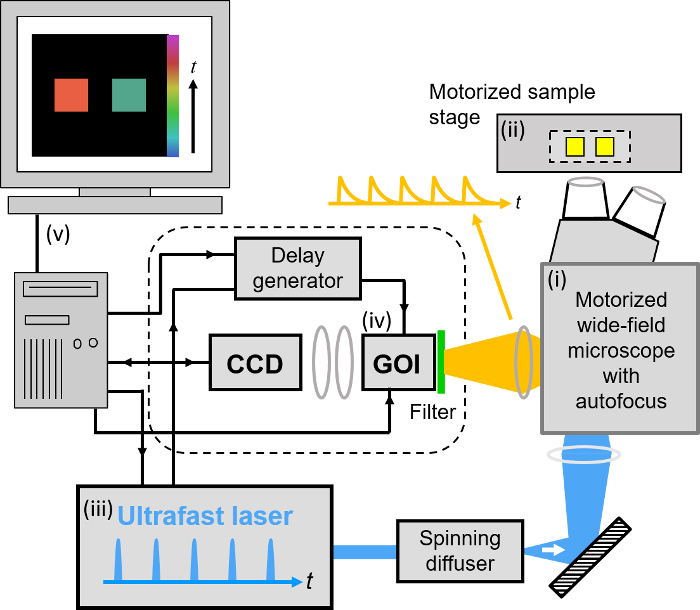
Figure 2. Schematic of wide-field openFLIM-HCA using a gated optical image intensifier (GOI). See main text for more detailed description of the system components. Please click here to view a larger version of this figure.
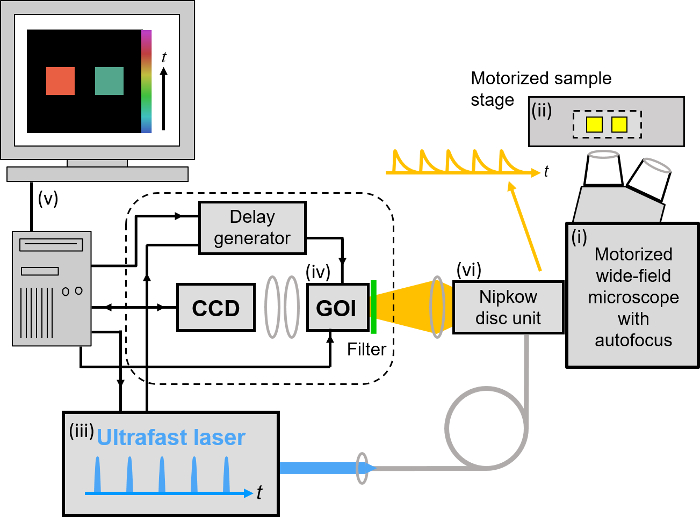
Figure 3. Schematic of optically sectioned openFLIM-HCA using a gated optical image intensifier (GOI) with a Nipkow disc scanner unit. See main text for more detailed description of the system components. Please click here to view a larger version of this figure.
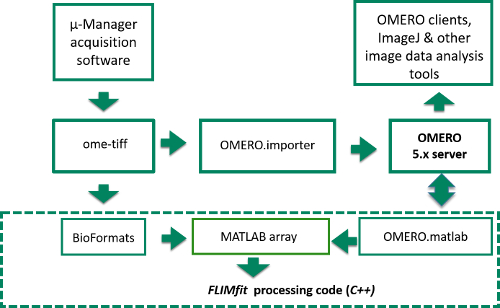
Figure 4. Schematic of image data flow from Manager acquisition program to OMERO server or computer disk and into FLIMfit for analysis. Data flow starts in top left corner where raw image data is acquired in μ-Manager acquisition software. The items inside the dashed box represent the stages of the FLIM data analysis, while those outside correspond to the data acquisition and storage. Please click here to view a larger version of this figure.
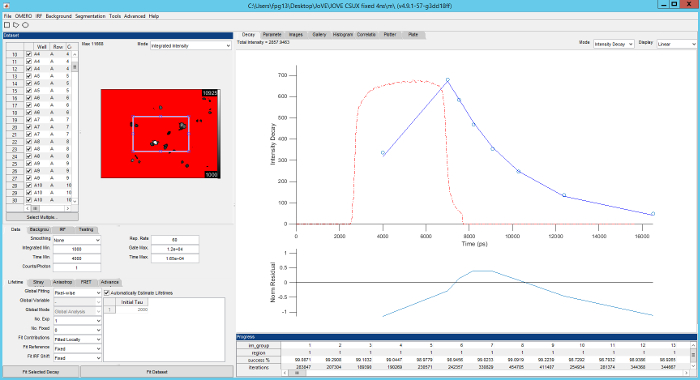
Figure 5. Screenshot of FLIMfit user interface. Top left, list of fields of view currently loaded and fluorescence intensity image of the currently selected field of view. Bottom left, selection of fitting model and fitting conditions. Top right, fluorescence decay for current region of interest (blue circles), fit to data (red line), IRF (red dashed line) with fit residuals below (blue line). Please click here to view a larger version of this figure.
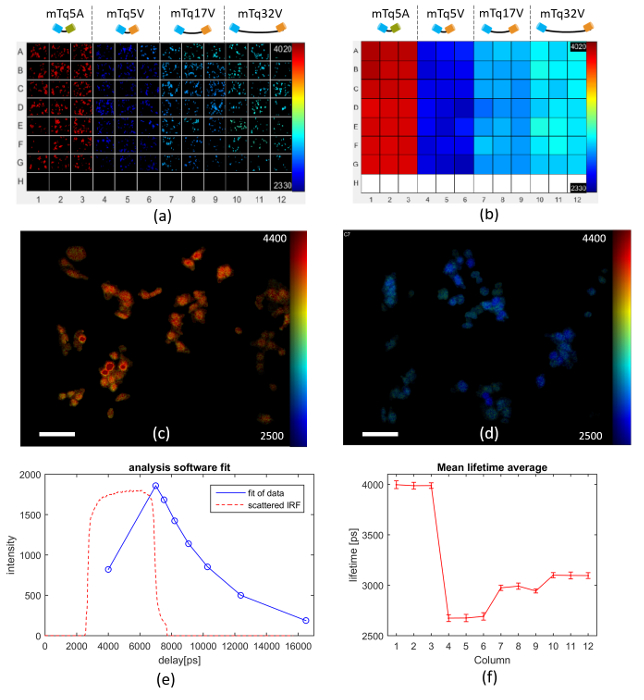
Figure 6. Multiwell plate FLIM of FRET model constructs mTq5V, mTq17V, mTq32V expressed in Cos cells. (a) exemplar fluorescence lifetime images of FOV in each well for different FRET constructs expressed in Cos cells as discussed in text. (b) heat map of mean Venus fluorescence lifetimes averaged over all cells in each well. (c) intensity image of mTurquoise Amber overlaid with lifetime map (scale bar 20 µm). (d) intensity image of mTurquoise Venus (17 amino acid) overlaid with lifetime map (scale bar 20 µm). (e) measured intensity decay profile (blue circles) of mTq5A construct (negative control) fluorescence averaged over all the cells imaged in well A1, together with the fit of the data to a monoexponential decay (blue solid line) and IRF (dashed orange line). (f) graph of mean lifetime averaged over each column across the multiwell plate, with error bars showing the standard deviation in fluorescence lifetime between fields of view. For (a-d) lifetime values are represented using a false color scale with the indicated limits in picoseconds. Please click here to view a larger version of this figure.
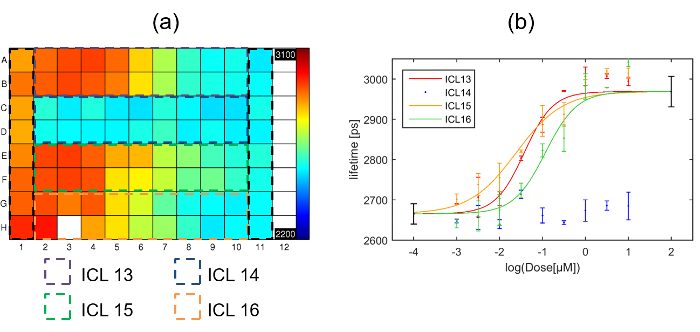
Figure 7. Exemplar multiwell plate FLIM screen of Gag inhibitors using HeLa cells expressing Gag-CFP. (a) Heat map of mean donor fluorescence lifetime per well for multiwell plate arrayed with column 1 presenting cells transfected with myr-Gag-CFP as a positive control for inhibition, column 11 presenting cells transfected with Gag-CFP exposed to vehicle only, and the dashed colored squares indicating the wells that were dosed with one of the four different inhibitors with concentrations ranging from high dose column 2 to low dose column 10. The false color scale represents the mean fluorescence lifetime ranging from 2,200 ps to 3,100 ps. (b) plots fluorescence lifetime for each inhibitor with error bars showing the standard deviation between repeat wells. Points in black at 2 and -4 on the horizontal axis are the positive and negative control points respectively obtained from columns 1 and 11 respectively. The solid lines indicate a fit to the dose response curve data for the different inhibitors. Please click here to view a larger version of this figure.
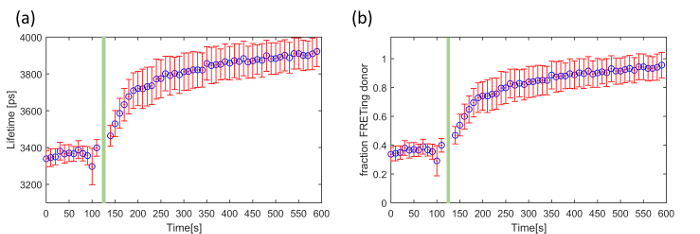
Figure 8. Exemplar use of FLIM to read out the EPAC FRET biosensor in a time-lapse measurement using HEK293T cells. Change in (a) mean fluorescence lifetime and (b) fraction of FRETing donor as a function of time following addition of forskolin indicated by the green bar. The error bars show the standard error per cell. Please click here to view a larger version of this figure.
| per Well | STD | ERR | per Well | STD | ERR | |||
| mT5A | 4008 | 32 | 12 | mT17V | 3004 | 26 | 10 | |
| mT5V | 2717 | 35 | 13 | mT32V | 3133 | 30 | 11 |
Table 1. Mean lifetime and standard deviation (STD) of the FRET constructs.
