The main attribute of intravital imaging is the longitudinal visualization of cellular processes without invasive intervention. For this, transgenic animals expressing a constitutive or inducible fluorescent marker in cells of interest are used. Figure 2 illustrates the expression of fluorescent labels in eNOStag-green fluorescent protein (GFP)9 and Rosa-mTmG mouse lines10. The eNOStag-GFP is a mouse line that was produced in-house using a truncated eNOS gene as a tag for GFP. Importantly, eNOS is highly expressed in endothelial cells, and the GFP signal is clearly seen in th eGolgi and cellular membrane (Figure 2A). Rosa-mTmG mice have a membrane-targeted two-color fluorescence Cre reporter allele. This line expresses mTomato fluorescence in widespread cells and mGFP in Cre recombinase-expressing cells and is vastly used for lineage tracing. As a tumor piece is transplanted from a non-transgenic donor, red fluorescence is predominantly expressed by stromal parts in the tumor (e.g., in the membrane of vascular cells, circulating cells, infiltrated blood cells, and tumor-associated fibroblasts (Figure 2B)).
Vessel formation is tightly regulated, and an excellent model to study this is the developing retina12,13. Also, tumor vessel growth is a kinetic process that is similar in principle to retinal vessel development. However, tumor vessels lack organization and, as a tumor is continuously remodeling, so is the tumor-associated vasculature. This can have its advantages when using the tumor as a angiogenic model, as all stages of vessel development can often be found in the same tumor at the same time. These include an endothelial sprouting front growing into an un-vascularized part of the tumor (Figure 2C); damaged vessels (Figure 2D); and an established vascular bed (Figure 2E-I) with mature, branched vessels (Figure 2E-II). However, angiogenic endothelial cells can also be found in these areas. When stimulated by angiogenic stimuli, an endothelial cell protrudes filopodia (Figure 2E-III) and can advance into a tip cell, using these filopodia for scanning and directional migration (Figure 2E-IV). This tip cell migrates into the tumor interstitium and is followed by dividing stalk cells. A single endothelial tip cell has several filopodia expanding, retracting and renewing at any time, and can be tracked using intravital microscopy (Figure 2F).
Secondly, intravital microscopy can be used to determine lumen formation at an angiogenic front, blood flow throughout the tumor, and extravasation. Depending on already-present intrinsic fluorescent signals in the animal, different fluorescent compounds can be administered. Blood flow and extravasation can be visualized using Hoechst, fluorescent dextrans, or FITC-BSA. For extended studies on blood flow and particle extravasation, long circulation fluorescently labeled pegylated nanoparticles of 100 nm can be used. For details on the preparation of these nanoparticles, see a previous publication5. The employment of these compounds is demonstrated in Figure 3. The endothelial tip cells in a Rosa-mTmG mouse can be clearly seen invading the tumor tissue. Functional blood flow is demonstrated by the purple fluorescence of systemically injected nanoparticles in the lumen of vascular tubes (Figure 3A-I). Nanoparticles closely reach endothelial tip cells illustrating the formation of a lumen in the stalk cell area (Figure 3A-II). The delivery of systemically administered chemotherapeutics depends on a functional vasculature in order to reach the tumor interstitium, and as shown in Figure 3B, injectable agents, independent of their size, do not penetrate areas with destroyed vessels, compressed vessels, or vessels with blood stasis.
In most organs, the endothelial lining forms a functional barrier between the blood and underlying tissue, and the passage of the molecules is tightly regulated. Tumor-associated vasculature, however, is known to be leaky, and pore-size cut-off depends greatly on the tumor type14. Fluorescent-conjugated dextrans with different sizes can be injected to assess the blood flow, permeability, and extravasation. Hoechst (615 Da) diffuses rapidly into tumor tissue and is taken up by surrounding cells (Figure 3C-I). Shortly after the injection, dextran of 10 KDa (Figure 3CII) and 2 MDa (Figure 3C-III) are found in the blood. However, dextran 10 KDa is also seen in the tumor interstitium, indicating the permeability of the endothelial lining for smaller molecules, which is a feature ascribed to tumor vessels. 40 min after injection (Figure 3C-IV), dextran 10 KDa is cleared from the blood stream (Figure 3C-V), and the fluorescent intensity of dextran 2MDa is decreased as well (Figure 3C-VI). However, large dextrans are not found in the tumor interstitium, demonstrating a lack of permeability to large molecules within this time frame.
Tumor pathophysiology, with its highly proliferating, necrotic, and un-vascularized areas, can be quite informative when using the tumor as an angiogenic model. However, this presents a problem for effective therapy and investigation. The heterogeneity of tumor-associated vasculature causes a heterogeneous distribution of administered drugs, leaving entire areas drug-free15. To improve drug delivery, several strategies can be applied15,16,17, and therapeutic progression can be examined using this intravital design. The tumor-associated vasculature can be manipulated using vasoactive agents to improve drug delivery17. The results of tumor vessel manipulation using tumor necrosis alpha (TNF) as a vasoactive agent in combination with Doxil, the encapsulated liposomal formulation of doxorubicin, as a chemotherapeutic agent are presented in this manuscript5,18,19. In contrast to dextrans, pegylated nanoparticles are made to circulate for several hours, if not days, in the blood circulation20.
As previously mentioned, the tumor area can be pre-scanned to identify the correct tumor areas depending on the research question. Drug fate can be evaluated in areas with a functional vasculature versus areas with an already damaged vascular bed (data not shown). To investigate the fate of chemotherapeutic agents without cytotoxic interference, nanoparticles with the same composition as the therapeutic agent can be used as a model drug. An eNOStag-GFP animal was treated i.v. with these nanoparticles and imaged 24 h later. Nanoparticles were still present in the vasculature, with minimal extravasation into the tumor interstitium (Figure 4A-I). However, when nanoparticles were administered in combination with TNF, extravasation was observed from the blood stream into the tumor interstitium (Figure 4A-II), increasing intratumoral drug delivery. By using high-resolution objective lenses, the intracellular localization of a compound can be recognized, as shown here by the nuclear location of Hoechst in green endothelial cells and cells of the tumor interstitium (Figure 4B). Moreover, as many chemotherapeutic agents, like doxorubicin, intercalate with DNA, the localization of these compounds can be evaluated. Doxorubicin has red fluorescent properties, and the nanocarrier can be labeled with, for instance, tetramethylindotricarbocyanine perchlorate (DiD). An eNOStag-GFP animal was injected i.v. with Doxil-DiD in combination with TNF, and images were taken 24 h after treatment. Doxil-DiD extravasated out of the blood vessels and was taken up by surrounding tumor tissue (Figure 4C-I). A more detailed evaluation of individual cells (Figure 4C-II) showed that the carrier can be found in the cytoplasm, while released doxorubicin was observed in the cellular nucleus.

Figure 1: Instruments, dorsal skinfold chamber, and equipment setup needed for the procedure. (A) Tumor fragments (a) and surgical instruments needed for implantation. Non-standard instruments are an ear puncher (b), a micro screw driver (c), and a bent needle (d). (B) Dorsal skinfold window chamber. Front (a) and back (b) window (arrow: holes for sutures; arrowhead: holes for bolts), 2 bolts (c), 2 nuts (d), 1 filler glass (e), 2 cover glasses (f), and 2 retaining rings (g). Also, retaining rings without hooks (white arrowhead) can be used when required. (C) Custom-made chamber holder (a), bolts for the chamber-to-chamber holder (b), temperature-controlled platform (c), bolts to secure the chamber holder to the platform (d), and the holder with the anesthesia tube (e). (D) Animal mounted in the chamber holder on the platform. A B16BL6 tumor (arrow) is visible in the window view area. (E) Equipment needed for the intravital evaluation. Computer with imaging and microscope control software (a), standard fluorescent light (b), and the microscope. A multiphoton confocal was used (c) with an anesthesia unit (d). Please click here to view a larger version of this figure.
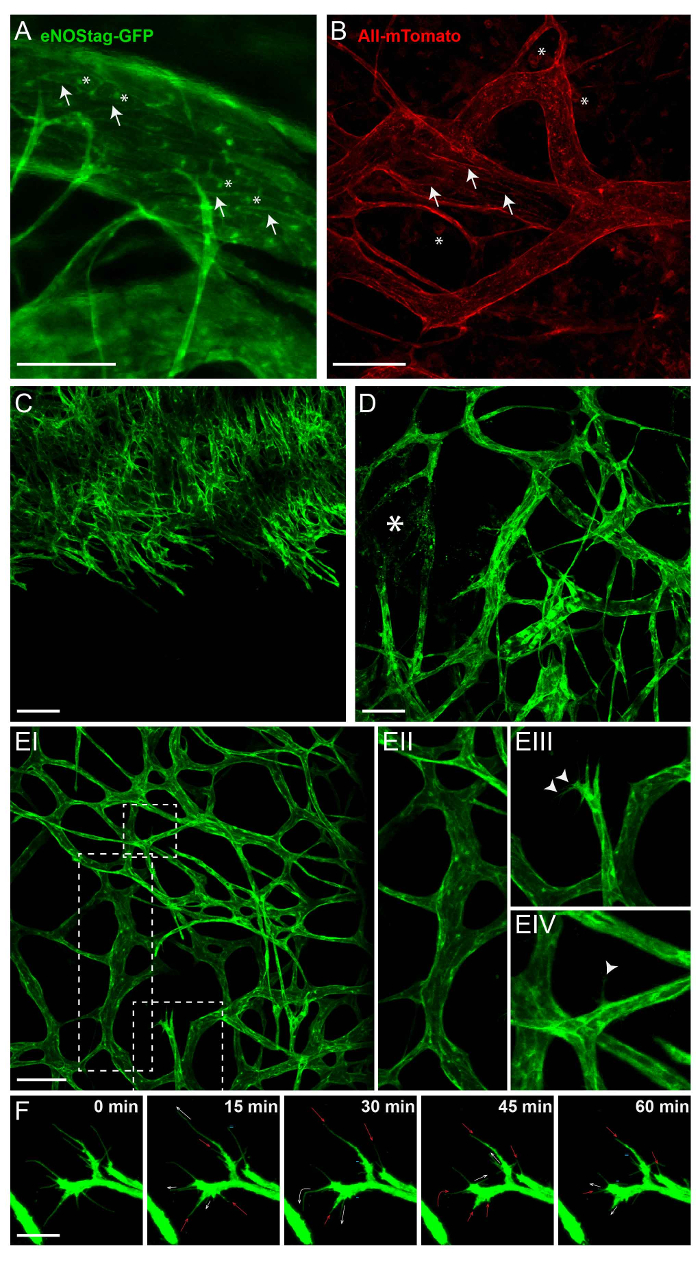
Figure 2: Intrinsic fluorescence of transgenic animals. All images shown here are Z-stack projections between 50 and 70 µm thick. (A) In the eNOSGFP-tag line, GFP is expressed in the Golgi (asterisk) and cell membrane (arrow) of endothelial cells. Scale bar = 100 µm. (B) Intratumoral expression of mTomato in the Rosa-mTmG line is predominantly found in the cell membrane of vascular cells (arrow) and blood cells (asterisk). Scale bar = 100 µm. (C) A projection of a growing angiogenic front into un-vascularized tumor. Scale bar = 100 µm. (D) A projection of part of the tumor with established and damaged vessels (asterisk). (E) A projection of established tumor vasculature (EI) showing mature tick vessels (EII); angiogenic endothelial tip cells with filopodia (EIII, arrowhead); and a mature vessel, from which a single endothelial cell extends a filopodium (EIV, arrowhead) into the interstitium. Scale bar = 100 µm. (F) Movement of the filopodia, followed for 1 h. Every 15 min, a Z-stack was taken; a maximum projection is presented here. The filopodia are extending (white arrow), stagnant (=), retracting (red arrow), or even completely disappearing (-). Scale bar = 25 µm. Please click here to view a larger version of this figure.
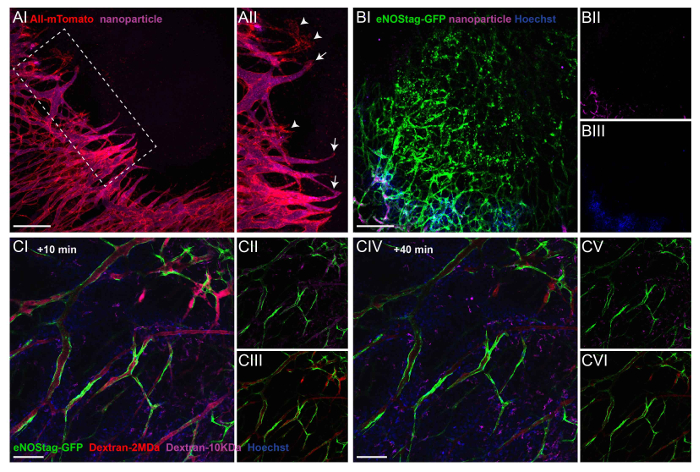
Figure 3: Administration of injectable labels to illustrate extravasation and blood flow. (A) A Rosa mTmG mouse was injected with purple fluorescent nanoparticles, and a Z-stack of an invading tip cell front was taken 10 min later (AI). A projection showing that most endothelial sprouts have a functional lumen (AII, arrow). Only rarely can endothelial sprouts be seen without flow (AII, arrowhead). Scale bar = 250 µm. (B) This Z-stack projection shows a destroyed tumor vessel area in an eNOStag-GFP animal prior to treatment (BI). The animal was injected with nanoparticles (purple, BII) and Hoechst (blue, BIII). Nanoparticles and Hoechst do not reach destroyed areas, indicated by granulated cell debris still expressing GFP. Scale bar = 250 µm. (C) An eNOStag-GFP mouse was injected with two dextrans of different sizes (red = Dextran 2 MDa; purple = dextran 10 KDa) and Hoechst (blue = 615 Da), and single-plane images are presented here. 10 min after the injection, presence in the blood and extravasation are seen in the same image. Hoechst extravasates almost immediately out of the blood vessels and is taken up by the surrounding cells (CI). Dextran 10 KDa (CII) can be seen in vessels and in the tumor interstitium. Dextran 2 MDa (CIII) can be found in the vessels. 40 min after injection (CIV), Dextran 10 KDa disappears from the blood (CV), and the fluorescent intensity of Dextran 2 MDa was also diminished (CVI). Scale bar = 100 µm. Please click here to view a larger version of this figure.
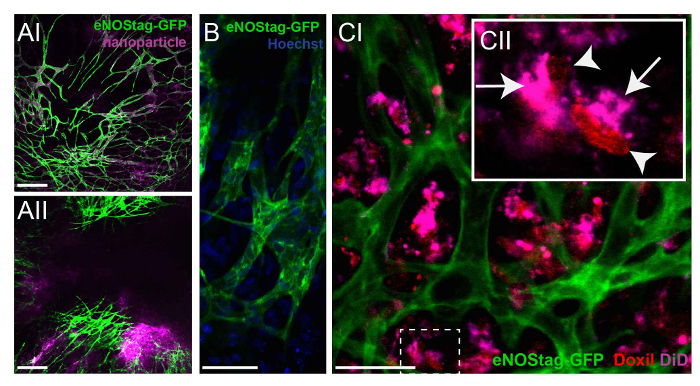
Figure 4: Investigating the fate of chemotherapeutic agents. (A) An eNOStag-GFP animal was treated i.v. with nanoparticles and imaged 24 h later. Nanoparticles are still present in vessels, with minimal extravasation in the tumor interstitium (AI). When nanoparticles are combined with TNF, extravasation is observed from the blood stream into the tumor interstitium (AII). Scale bar = 250 µm. (B) Using high-resolution lenses, the endothelial cytoplasm (green) and nucleus (blue) of an individual cell can be recognized. Scale bar = 50 µm. (C) An eNOStag-GFP animal was injected i.v. with Doxil-DiD in combination with TNF, and images were taken 24 h later. Here, the extravasation of Doxil-DiD out of the vessels into the tumor interstitium is obvious (CI). A more detailed evaluation of individual cells (CII) shows that the purple carrier can be found in the cytoplasm (arrow), while the released doxorubicin (red) is observed in the cellular nucleus (arrowhead). Scale bar = 50 µm. Please click here to view a larger version of this figure.
