Synthesis of a Water-soluble Metal–Organic Complex Array
Summary
A potential general method for the synthesis of water-soluble multimetallic peptidic arrays containing a predetermined sequence of metal centers is presented.
Abstract
We demonstrate a method for the synthesis of a water-soluble multimetallic peptidic array containing a predetermined sequence of metal centers such as Ru(II), Pt(II), and Rh(III). The compound, named as a water-soluble metal-organic complex array (WSMOCA), is obtained through 1) the conventional solution-chemistry-based preparation of the corresponding metal complex monomers having a 9-fluorenylmethyloxycarbonyl (Fmoc)-protected amino acid moiety and 2) their sequential coupling together with other water-soluble organic building units on the surface-functionalized polymeric resin by following the procedures originally developed for the solid-phase synthesis of polypeptides, with proper modifications. Traces of reactions determined by mass spectrometric analysis at the representative coupling steps in stage 2 confirm the selective construction of a predetermined sequence of metal centers along with the peptide backbone. The WSMOCA cleaved from the resin at the end of stage 2 has a certain level of solubility in aqueous media dependent on the pH value and/or salt content, which is useful for the purification of the compound.
Introduction
Controlled synthesis of complicated molecular structures has always been a major issue in synthetic chemistry. From this point of view, to synthesize multinuclear heterometallic complexes in a designable fashion is still a worthy subject to be challenged in the field of inorganic chemistry because of the numbers of possible structural outcomes from the ligand-metallation-based approach that is commonly used for the preparation of monomeric metal complexes. Although several examples of multinuclear heterometallic complexes have been reported so far1,2,3, the trial-and-error or arduous nature of their synthesis necessitates the development of a simple method that is applicable for a wide range of structures.
As a new approach to address this issue, in 2011 we reported a synthetic methodology4,5 where various mononuclear metal complexes having a Fmoc-protected amino acid moiety are sequentially coupled to give multimetallic peptidic arrays by using the protocols of solid-phase polypeptide synthesis6. Due to the consecutive nature of polypeptide synthesis, a specific sequence of multiple metal centers is rationally designable by controlling the number and order of the coupling reactions of those metal complex monomers. Later, this approach was further modularized to make various larger and/or branched array structures by combining with the covalent linkage between two shorter arrays7.
Here we will show how the synthesis of such multimetallic peptidic arrays is typically operated by choosing the recently reported WSMOCA (18 CAS RN 1827663-18-2; Figure 1) as a representative example. Although the synthesis of one particular array is described in this protocol, the same procedures are applicable to the synthesis of a wide range of different sequences, including isomers9. We expect that this protocol will inspire more researchers to participate in the science of sequence-controlled compounds, where the molecules investigated thus far have typically been biopolymers but rarely include examples of metal-complex-based species.
Protocol
1. Preparation of Metal Complex Monomers (2 CAS RN 1381776-70-0, 3 CAS RN 1261168-42-6, 4 CAS RN 1261168-43-7; Figure 1)
- Preparation of Ru monomer 2
- Combine the organic precursor (59 CAS RN 1381776-63-1; Figure 1) (380 mg, 0.48 mmol) and [Ru(p-cymene)Cl2] dimer (224 mg, 0.37 mmol) with a stir bar in a 100 ml single-neck round-bottom flask.
- Add methanol (MeOH) (25 ml) to the mixture, connect a condenser to the joint of the flask, and stir the suspension at 65 °C for 3 hr in a temperature-controlled oil bath.
- Cool the reaction mixture to room temperature and filter the suspension through a filter paper by suction.
- Wash the residue on the filter thoroughly with MeOH until the filtrate becomes visually colorless and dry the residue under reduced pressure.
- Combine the residue and 4'-(4-methylphenyl)-2,2':6',2"-terpyridine (216 mg, 0.68 mmol) with a stir bar in a 100 ml single-neck round-bottom flask.
- Add MeOH (22.5 ml) and water (2.5 ml) to the mixture, connect a condenser to the joint of the flask, and stir the suspension at 70 °C for 16 hr.
- Cool the reaction mixture to room temperature and filter the suspension.
- Dry the residue on the filter under reduced pressure and dissolve it in dimethyl sulfoxide (DMSO) (3 ml).
- Add the DMSO solution slowly to an excess of ethyl acetate (EtOAc).
- Filter the resultant suspension, wash the residue on the filter with EtOAc, and dry it under reduced pressure.
- Preparation of Pt monomer 3
- Combine the organic precursor (64 CAS RN 1261168-39-1; Figure 1) (360 mg, 0.50 mmol) and Pt(cycloocta-1,5-diene)Cl2 (195 mg, 0.52 mmol) with a stir bar in a 100 ml single-neck round-bottom flask.
- Add MeOH (15 ml) to the mixture, connect a condenser to the joint of the flask, and stir the suspension at 65 °C for 12 hr.
- Cool the reaction mixture to room temperature and filter the suspension.
- Wash the residue on the filter thoroughly with MeOH and dry it under reduced pressure.
- Preparation of Rh monomer 4
- Combine the organic precursor (6; Figure 1) (360 mg, 0.50 mmol) and RhCl3·3H2O (137 mg, 0.52 mmol) with a stir bar in a 100 ml single-neck round-bottom flask.
- Add MeOH (50 ml) to the mixture, connect a condenser to the joint of the flask, and stir the suspension at 65 °C for 12 hr under a N2 atmosphere.
- Cool the reaction mixture to room temperature and filter the suspension.
- Wash the residue on the filter thoroughly with MeOH and dry it under reduced pressure.
2. Preparation of Water-soluble Metal–Organic Complex Array 1
- Fmoc Deprotection from TG Sieber Resin
- Combine as-purchased TG Sieber resin (135 mg) with a stir bar in a 10 ml 2-neck flask bearing a drain at the bottom equipped with a glass filter and a 2-way stop cock (Figure 2a). Connect a 3-way stop cock and a glass stopper to the joints of the flask.
- Exchange the internal atmosphere with N2 by using a vacuum line, and then swell the resin with anhydrous-grade dichloromethane (CH2Cl2) (1 ml) (Figure 2b).
- Add anhydrous-grade dimethylformamide (DMF) (3 ml) and piperidine (1 ml) in this order and stir the mixture for 2.5 hr at room temperature.
- Remove the solution by filtration through the drain. Wash the resin with anhydrous-grade MeOH (3 ml, 3 min stirring) and anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) alternately three times and then with anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) four times (Figure 2c).
- Combine all the solutions obtained in 2.1.4 and dilute them with acetonitrile (CH3CN) to a volume of 50 ml. Transfer an aliquot (1 ml) of the resultant solution into a quartz cuvette with an optical length of 1 cm and dilute it with CH3CN (2 ml).
- Determine the mol number of deprotected Fmoc moiety (f µmol) based upon the extinction coefficient of piperidine-dibenzofulvene adduct (6,234 at 299 nm)10 and the spectroscopically obtained absorbance (a) of the solution prepared through protocol 2.1.5 according to the following equation:
f = 0.05 x 106 x 3 x a/6234
- Loading of Ru monomer 2
- Add anhydrous-grade CH2Cl2 (2.5 ml), Ru monomer 2 (64.9 mg, 53.2 µmol), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (30.3 mg, 79.8 µmol), anhydrous-grade DMSO (2.5 ml), and N,N-diisopropylethylamine (iPr2NEt) (20 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature (Figure 2d).
- Remove the solution by filtration through the drain. Wash the resin with anhydrous-grade DMSO (3 ml, 5 min stirring) three times, anhydrous-grade MeOH (3 ml, 3 min stirring) and anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) alternately three times, and anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) three times.
- Add anhydrous-grade CH2Cl2 (5 ml), benzoic anhydride (0.28 g, 1.5 mmol), and N-methylimidazole (0.10 ml, 1.5 mmol) in this order under a N2 atmosphere to the washed resin and stir the mixture for 2 hr at room temperature.
- Remove the solution by filtration through the drain. Wash the resin with anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) and anhydrous-grade MeOH (3 ml, 3 min stirring) alternately three times and then with anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) three times.
- Repeat the protocols as described in 2.1.3-2.1.6 to quantify the mol number of loaded Ru monomer 2.
- Loading of Fmoc- and side-residue tertiary-butyl (tBu)-protected (L)-glutamic acid (Glu) (7 CAS RN 71989-18-9; Figure 1)
- Add anhydrous-grade CH2Cl2 (4.5 ml), Glu·H2O (39.4 mg, 88.8 µmol), HBTU (50.5 mg, 133.2 µmol), anhydrous-grade DMSO (0.5 ml), and iPr2NEt (50 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature (Figure 2e).
NOTE: The amounts of Glu·H2O and HBTU are gradually decreased from steps 2.3 to 2.7 to keep their stoichiometry to the reactive -NH2 functionality on the resin constant. - Repeat the protocols as described in 2.2.2-2.2.4.
- Take a small portion of the resin out of the flask and put it into a mixture of trifluoroacetic acid (CF3CO2H) (2.5 µl), triethylsilane (Et3SiH) (0.5 µl), and 1,2-dichloroethane (47 µl). Sonicate the mixture for 0.5 hr and use the resultant solution for mass spectrometry4,7,8,9 (Figure 3a).
- Repeat the protocols as described in 2.1.3-2.1.6 to quantify the mol number of loaded Glu.
- Add anhydrous-grade CH2Cl2 (4.5 ml), Glu·H2O (39.4 mg, 88.8 µmol), HBTU (50.5 mg, 133.2 µmol), anhydrous-grade DMSO (0.5 ml), and iPr2NEt (50 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature (Figure 2e).
- Loading of Pt monomer 3
- Add anhydrous-grade DMSO (4.5 ml), Pt monomer (32.9 mg, 33.3 µmol), HBTU (18.9 mg, 50.0 µmol), anhydrous-grade CH2Cl2 (0.5 ml), and iPr2NEt (20 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature (Figure 2f).
- Repeat the protocols as described in 2.2.2-2.2.5 to quantify the mol number of loaded Pt monomer 3.
- Loading of Glu
- Add anhydrous-grade CH2Cl2 (4.5 ml), Glu·H2O (27.8 mg, 62.9 µmol), HBTU (35.8 mg, 94.4 µmol), anhydrous-grade DMSO (0.5 ml), and iPr2NEt (50 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature.
- Repeat the protocols as described in 2.3.2-2.3.4 (Figure 3b).
- Loading of Rh monomer 4
- Add anhydrous-grade DMSO (4.5 ml), Rh monomer 4 (21.8 mg, 23.3 µmol), HBTU (13.3 mg, 35.0 µmol), anhydrous-grade CH2Cl2 (0.5 ml), and iPr2NEt (20 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature (Figure 2g).
- Repeat the protocols as described in 2.2.2.
- Repeat the protocols as described in 2.6.1.
- Repeat the protocols as described in 2.2.2-2.2.5 to quantify the mol number of loaded Rh monomer 4.
- Loading of Glu
- Add anhydrous-grade CH2Cl2 (4.5 ml), Glu·H2O (20.4 mg, 46.0 µmol), HBTU (26.2 mg, 69.0 µmol), anhydrous-grade DMSO (0.5 ml), and iPr2NEt (50 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature.
- Repeat the protocols as described in 2.3.2-2.3.4 (Figure 3c).
- Loading of 2-[2-(2-methoxyethoxy)ethoxy]acetic (TEG) acid (8 CAS RN 16024-58-1; Figure 1)
- Add anhydrous-grade CH2Cl2 (3 ml), TEG acid (14 µl, 91.0 µmol), HBTU (51.7 mg, 136.5 µmol), anhydrous-grade CH2Cl2 (2 ml), and iPr2NEt (50 µl) in this order under a N2 atmosphere to the washed resin and stir the mixture for 12 hr at room temperature.
- Remove the solution by filtration through the drain. Wash the resin with anhydrous-grade DMSO (3 ml, 5 min stirring) two times, anhydrous-grade MeOH (3 ml, 3 min stirring) and anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) alternately three times, and anhydrous-grade CH2Cl2 (3 ml, 3 min stirring) three times.
- Cleavage from the resin at the end of solid-phase synthesis
- Wash the resin with diethyl ether (4 ml, 5 min stirring) three times, dry it under a vacuum, and swell it with anhydrous-grade CH2Cl2 (1 ml, 5 min stirring).
- Add a mixture of CF3CO2H (0.1 ml), Et3SiH (20 µl), and 1,2-dichloroethane (1.9 ml) to the suspension and stir the mixture for 12 hr at room temperature.
- Remove the solution by filtration through the drain, add a new mixture of CF3CO2H (0.1 ml), Et3SiH (20 µl), and 1,2-dichloroethane (1.9 ml) to the resin, and stir the mixture for 1 hr at room temperature.
- Repeat step 2.9.3 until the solution becomes visually colorless (Figure 2h).
- Combine all the solutions as obtained through protocols 2.9.2-2.9.4 and analyze the contents of the resultant solution by mass spectrometry4,7,8,9 (Figure 3d).
- Remove volatile species of the solution by evaporation and dissolve the residue in a mixture of CF3CO2H (0.2 ml), Et3SiH (40 µl), and 1,2-dichloroethane (3.8 ml).
- Stir the mixture for 24 hr at room temperature and analyze the contents of the resultant solution by mass spectrometry4,7,8,9 to confirm the complete deprotection of tBu groups at the side residues of 1 (Figure 3e).
- Remove volatile species of the solution by evaporation.
- Purification of 1
- Sonicate the solid residue as obtained through protocol 2.9 in CH2Cl2 and decant the solution. Repeat this process until the decant solution becomes visually colorless.
- Analyze the contents of the resultant solid residue by mass spectrometry4,7,8,9.
- Wash the residue with MeOH (100 µl/10 mg) with sonication, decant the solution, and analyze the contents of the resultant solid residue by mass spectrometry4,7,8,9.
- Dissolve the residue (1 mg) in a mixture of CH3CN (90 µl) and water (10 µl) by sonication and combine the resultant solution with phosphate-buffered saline (800 µl, 10 mM, pH = 7.4). Sonicate the mixture and incubate it at 37 °C for 24 hr.
- Extract the colored species in the decant supernatant with a mixture of 1,2-dichloroethane (500 µl), CH3CN (20 µl), and CF3CO2H (20 µl). Analyze the extract by mass spectrometry4,7,8,9 (Figure 3f).
- Repeat the extraction until the aqueous phase becomes visually colorless. Combine the organic solutions and remove volatile species by evaporation.
Representative Results
Figure 1 shows the molecular structures of the final target compound, precursors, and intermediates. Figure 2 shows the images of the resin and Figure 3 shows the MALDI-TOF mass spectra of samples at selected procedure steps. Images from Figure 2a to 2h show the changes in the color and appearance of the resin that it undergoes during the reaction steps in section 2 of the protocol. MALDI-TOF mass spectrometry is used to trace the reactions and to confirm the presence of target species as expected.

Figure 1. Molecular structures of the WSMOCA, precursors, and intermediates. (1) the targeted WSMOCA; (2, 3, 4) the Ru, Pt, and Rh monomers, respectively; (5) the organic precursor for Ru monomer 2; (6) the organic precursor for Pt monomer 3 and Rh monomer 4; (7) Glu; (8) TEG acid; (9, 10, 11) synthetic intermediates to be detected at 2.3.3, 2.5.2, and 2.7.2, respectively. Please click here to view a larger version of this figure.

Figure 2. Appearances of the resin at the selected synthetic steps. Photos of (a) as-purchased TG Sieber resin in the glassware for solid-phase synthesis at 2.1.1; (b) the resin swelled in CH2Cl2 at 2.1.2; (c) the resin washed after the deprotection of Fmoc groups at 2.1.4; (d) the resin suspended in a solution for the loading of Ru monomer 2 at 2.2.1; (e) the resin suspended in a solution for the loading of Glu at 2.3.1; (f) the resin suspended in a solution for the loading of Pt monomer 3 at 2.4.1; (g) the resin suspended in a solution for the loading of Rh monomer 4 at 2.6.1; (h) the resin suspended in a solution for the cleavage reaction at 2.9.4. Please click here to view a larger version of this figure.
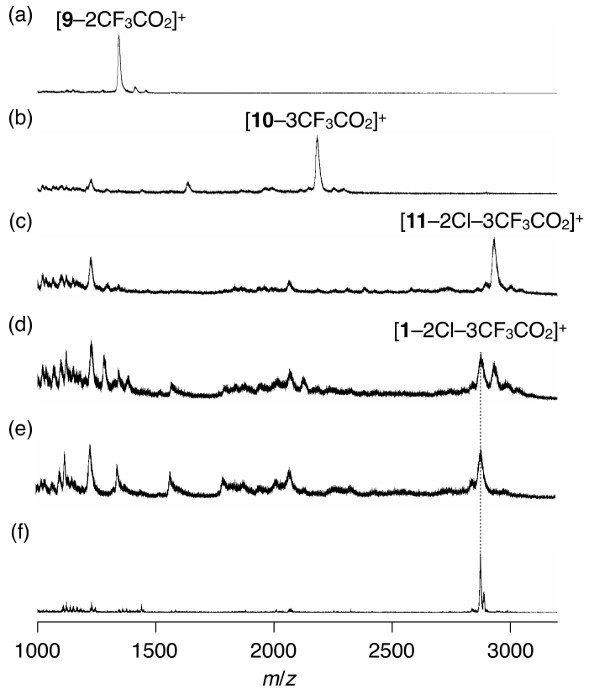
Figure 3. Traces of the preparation of 1 determined by mass spectrometric analysis. MALDI-TOF mass spectra of samples as-cleaved from the resin at the selected steps of the solid-phase synthesis (procedures (a) 2.3.3 to confirm the presence of 9; (b) 2.5.2 to confirm the presence of 10; (c) 2.7.2 to confirm the presence of 11; (d) 2.9.5 to confirm the presence of 1) and those of the samples at the following steps (procedures (e) 2.9.7 to confirm the complete deprotection of tBu groups at the side residues of 1; (f) 2.10.5 to confirm the absence of any major signals other than that of 1). Please click here to view a larger version of this figure.
Discussion
Perfect removal of the undesired chemicals from the resin is not always possible simply by washing with solvents that can easily dissolve those chemicals. A key technique to efficiently wash the resin is to cause it to swell and shrink repetitively so that the chemicals remaining inside will be forced out. This is why the resin in our procedure is treated with CH2Cl2 and MeOH alternately as it is washed (e.g., protocol 2.1.4).
As a consequence of successive multiple non-quantitative coupling reactions, the amount of targeted array in the as-cleaved mixture at the end of the solid-phase synthesis could be small. Although each reaction in the solid-phase synthesis is generally only conducted once, the same coupling reaction can be repeated multiple times, as exemplified in protocol 2.6, if it is necessary to improve the overall coupling yield of the corresponding reaction step. By repeating the same coupling reaction two times, a ~10% larger yield of the corresponding coupling reaction can be realized.
In contrast to Fmoc-protected amino acid monomers commonly used for solid-phase polypeptide synthesis, those monomers bearing a metal complex for multimetallic peptidic arrays generally show a yield of no more than 80% in their coupling reactions at the surface of the resin. Steric effects due to the presence of a bulky metal complex moiety play a role, as the insertion of one amino acid unit at the C-terminal of the monomer sometimes improves its coupling yield drastically. However, even such modifications of the monomer structure are still not enough to optimize the quantitative coupling reactions. This is an issue to be addressed in the future, particularly for the high-throughput production of multimetallic peptidic arrays via the automation of the whole process of this methodology, as already established in the case of solid-phase polypeptide synthesis.
Compared with solution-phase synthesis, one of the important advantages of solid-phase synthesis is the easy separation of products attached to the resin from other chemicals in the solution by filtration and washing with solvents that can dissolve them.11 This is particularly useful for the synthesis of multimetallic species whose separation/purification is not easy with other methods. Accordingly, the protocol highlighted here is the only realistic choice to make multimetallic peptidic arrays having a predetermined sequence of three or more different metals. Moreover, due to the simplicity of this method, the protocol can cover the production of a much wider range of multimetallic heteronuclear complexes than those accessible from already-existing synthetic approaches1,2,3.
As compounds produced by this method possess a perfectly controlled sequence of metal centers along the peptide backbone, they are appealing candidates to investigate the effects of such sequence-regulated structures on the interactions with bio-related molecules (e.g., peptides, proteins, nucleic acids, and sugars, which also have a regulated sequence in their structure). This is our incentive to make the products water-soluble.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the World Premier International Research Center (WPI) Initiative on Materials Nanoarchitectonics and a Grant-in-Aid for Challenging Exploratory Research (No. 26620139), both of which were provided from MEXT, Japan.
Materials
| Dichloro(p‐cymene)ruthenium(II), dimer | Kanto Chemical | 11443-65 | |
| Dichloro(1,5-cyclooctadiene)platinum(II) | TCI | D3592 | |
| Rhodium(III) chloride trihydrate | Kanto Chemical | 36018-62 | |
| Phosphate buffered saline, tablet | Sigma Aldrich | P4417-50TAB | |
| NovaSyn TG Sieber resin | Novabiochem | 8.55013.0005 | |
| HBTU | TCI | B1657 | |
| Benzoic anhydride | Kanto Chemical | 04116-30 | |
| Fmoc-Glu(OtBu)-OH・H2O | Watanabe Chemical Industries | K00428 | |
| Trifluoroacetic acid | Kanto Chemical | 40578-30 | |
| Triethylsilane | TCI | T0662 | |
| 2-[2-(2-Methoxyethoxy)ethoxy]acetic acid | Sigma Aldrich | 407003 | Dried over 3Å sieves |
| Dithranol | Wako Pure Chemical Industries | 191502 | |
| N-methylimidazole | TCI | M0508 | |
| N‐ethyldiisopropylamine | Kanto Chemical | 14338-32 | |
| Piperidine | Kanto Chemical | 32249-30 | |
| 4'-(4-methylphenyl)-2,2':6',2"-terpyridine | Sigma Aldrich | 496375 | |
| Dehydrated grade dimethylsulfoxide | Kanto Chemical | 10380-05 | |
| Dehydrated grade methanol | Kanto Chemical | 25506-05 | |
| Dehydrated grade N,N‐Dimethylformamide | Kanto Chemical | 11339-84 | Amine Free |
| Dehydrated grade dichloromethane | Kanto Chemical | 11338-84 | |
| MeOH | Kanto Chemical | 25183-81 | |
| Dimethylsulfoxide | Kanto Chemical | 10378-70 | |
| Ethyl acetate | Kanto Chemical | 14029-81 | |
| Acetonitrile | Kanto Chemical | 01031-70 | |
| 1,2-dichloroethane | Kanto Chemical | 10149-00 | |
| Diethyl ether | Kanto Chemical | 14134-00 | |
| Dichloromethane | Kanto Chemical | 10158-81 |
References
- Takanashi, K., et al. Heterometal Assembly in Dendritic Polyphenylazomethines. Bull. Chem. Soc. Jpn. 80, 1563-1572 (2007).
- Packheiser, R., Ecorchard, P., Rüffer, T., Lang, H. Heteromultimetallic Transition Metal Complexes Based on Unsymmetrical Platinum(II) Bis-Acetylides. Organometallics. 27, 3534-3536 (2008).
- Sculfort, S., Braunstein, P. Intramolecular d10-d10 Interactions in Heterometallic Clusters of the Transition Metals. Chem. Soc. Rev. 40, 2741-2760 (2011).
- Vairaprakash, P., Ueki, H., Tashiro, K., Yaghi, O. M. Synthesis of Metal-Organic Complex Arrays. J. Am. Chem. Soc. 133, 759-761 (2011).
- Jacoby, M. Synthesis: Method Couples Various Metals in Predetermined Sequences. C&EN. 89, (2011).
- White, P., Eds Dörner, B. Synthetic Notes. Peptide Synthesis 2008/2009. , (2009).
- Sajna, K. V., Fracaroli, A. M., Yaghi, O. M., Tashiro, K. Modular Synthesis of Metal-Organic Complex Arrays Containing Precisely Designed Metal Sequences. Inorg. Chem. 54, 1197-1199 (2015).
- Sukul, P. K., et al. A Water-Soluble Metal-Organic Complex Array as a Multinuclear Heterometallic Peptide Amphiphile That Shows Unconventional Anion Dependency in Its Self-Assembly. Chem. Commun. 52, 1579-1581 (2016).
- Fracaroli, A. M., Tashiro, K., Yaghi, O. M. Isomers of Metal-Organic Complex Arrays. Inorg. Chem. 51, 6437-6439 (2012).
- Gude, M., Ryf, J., White, P. D. An Accurate Method for the Quantitation of Fmoc-Derivatized Solid Phase Supports. Letters in Peptide Science. 9, 203-206 (2002).
- Merrifield, R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 85, 2149-2154 (1963).
